Scalable Synthesis of 6-Fluoro-3-Hydroxy-2-Pyrazinecarboxamide for Antiviral Pharmaceutical Intermediates
Introduction to Advanced Pyrazine Chemistry
The pharmaceutical industry continuously demands more efficient pathways for synthesizing complex heterocyclic intermediates, particularly those serving as core structures for antiviral agents. Patent CN102603658A introduces a groundbreaking preparation method for 6-fluoro-3-hydroxyl-2-pyrazinoic acid amide, a critical building block in the development of next-generation therapeutics targeting viral infections. This technical insight report analyzes the proprietary four-step synthesis route that transforms 6-bromo-3-amino-2-pyrazinoic acid amide into the target fluorinated hydroxy-pyrazine derivative with exceptional efficiency. Unlike traditional methods that rely on hazardous reagents and multi-step purifications, this novel approach leverages a strategic amino protection-deprotection sequence coupled with a mild halogen exchange mechanism. The result is a process that not only enhances chemical yield but also aligns with modern green chemistry principles by minimizing waste and operational complexity.
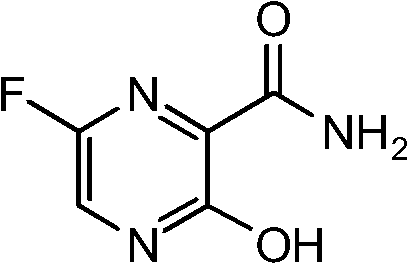
For R&D directors and process chemists, the significance of this patent lies in its ability to bypass the thermodynamic and kinetic barriers associated with direct nucleophilic substitution on electron-deficient pyrazine rings. By temporarily masking the reactive amino group with a tert-butoxycarbonyl (Boc) moiety, the synthesis prevents unwanted side reactions during the critical fluorination step. This level of molecular control ensures that the final product meets the stringent purity specifications required for GMP manufacturing. Furthermore, the use of common industrial solvents and reagents suggests that this pathway is not merely a laboratory curiosity but a viable candidate for immediate technology transfer and commercial scale-up.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated pyrazine derivatives has been plagued by inefficient routes that compromise both yield and safety. As detailed in the background technology of the patent, conventional methods often initiate from nitro-substituted precursors or require the conversion of di-chloro intermediates. A representative example of these legacy processes involves the use of phosphorus oxychloride (POCl3) under reflux conditions to introduce chlorine atoms, followed by harsh fluorination steps requiring strictly anhydrous environments. These conditions are not only energy-intensive but also pose significant safety risks due to the corrosive nature of the reagents involved. Moreover, the final conversion of a fluoro-substituent to a hydroxyl group in these old routes typically proceeds with dismal efficiency, often yielding less than 30% of the desired product after extensive purification.
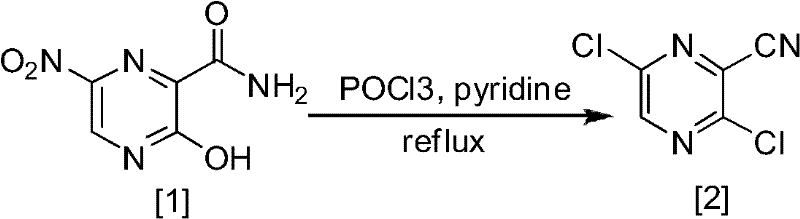
The reliance on column chromatography for purification in these traditional pathways further exacerbates the cost and time burden, making them unsuitable for large-scale industrial application. The cumulative effect of low yields across multiple steps results in a substantial loss of raw materials and a dramatic increase in the cost of goods sold. Additionally, the generation of hazardous waste streams from chlorinating agents and organic solvents creates significant environmental compliance challenges. For supply chain managers, these factors translate into unreliable delivery schedules and volatile pricing, as the complexity of the synthesis leaves little room for error or optimization.
The Novel Approach
In stark contrast, the method disclosed in CN102603658A offers a streamlined alternative that fundamentally reimagines the synthetic logic. By selecting 6-bromo-3-amino-2-pyrazinoic acid amide as the starting material, the inventors have identified a precursor that is both commercially accessible and chemically predisposed for selective modification. The core innovation lies in the sequential application of amino protection, halogen exchange, deprotection, and diazotization. This logical progression allows each functional group to be manipulated independently, avoiding the cross-reactivity issues that plague direct substitution methods. The bromine atom serves as an excellent leaving group for the subsequent fluorination, while the protected amino group remains inert until the final stages of the synthesis.
This novel approach eliminates the need for aggressive chlorinating agents like POCl3 and avoids the requirement for strictly anhydrous conditions in every step. The reaction conditions are mild, often proceeding at temperatures between 25°C and 80°C, which significantly reduces energy consumption. Furthermore, the workup procedures rely on standard extraction and crystallization techniques rather than expensive chromatographic separation. This shift from batch-wise purification to continuous isolation methods is a key enabler for commercial viability, ensuring that the process can be scaled from gram quantities in the lab to metric tons in the plant without losing efficiency or product quality.
Mechanistic Insights into Boc-Protection and Halogen Exchange
The success of this synthesis hinges on the precise execution of the Boc protection strategy in the first step. By reacting the starting amine with di-tert-butyl dicarbonate (Boc2O) in the presence of a base such as sodium hydride or triethylamine, the nucleophilicity of the amino group is effectively suppressed. This protection is crucial because free amines on the pyrazine ring can interfere with the subsequent nucleophilic aromatic substitution by coordinating with metal cations or undergoing unwanted side reactions. The formation of the 6-bromo-3-t-butyl carbamate-2-pyrazinoic acid amide intermediate creates a stable platform for the introduction of the fluorine atom. The steric bulk of the Boc group also helps to direct the incoming fluoride ion to the correct position on the ring, enhancing regioselectivity.
Following protection, the halogen exchange reaction utilizes a fluoride donor reagent, preferably potassium fluoride (KF), often assisted by a phase transfer catalyst like tetrabutylammonium bromide. In polar aprotic solvents such as DMSO or DMF, the fluoride ion becomes sufficiently nucleophilic to displace the bromine atom at the 6-position of the pyrazine ring. This SNAr mechanism is facilitated by the electron-withdrawing nature of the pyrazine ring and the adjacent amide group. The use of KF is particularly advantageous from a cost perspective compared to more exotic fluorinating agents like Selectfluor or DAST. The reaction proceeds cleanly to form the 6-fluoro-3-t-butyl carbamate intermediate, setting the stage for the final transformations.
Impurity control is inherently built into this mechanistic design. The distinct polarity differences between the protected intermediates and potential byproducts allow for easy removal via aqueous workups. For instance, unreacted starting materials or inorganic salts are efficiently washed away during the extraction phases described in the patent examples. The final diazotization step, which converts the deprotected amine into the hydroxyl group via a diazonium salt intermediate, is performed at low temperatures (-20°C to 10°C) to prevent decomposition. This careful temperature control ensures that the highly reactive diazonium species converts exclusively to the phenol-like hydroxyl product, minimizing the formation of tar or polymeric impurities that often contaminate such reactions.
How to Synthesize 6-Fluoro-3-Hydroxy-2-Pyrazinecarboxamide Efficiently
The synthesis of this high-value intermediate is achieved through a robust four-step protocol that balances reaction kinetics with operational simplicity. The process begins with the dissolution of the bromo-amino precursor in a polar solvent, followed by the controlled addition of the protecting group reagent. After isolation of the protected intermediate, the fluorination is conducted under heated conditions to drive the substitution to completion. The subsequent deprotection uses acidic hydrolysis to regenerate the amine, which is immediately subjected to diazotization in sulfuric acid.
- Protect the amino group of 6-bromo-3-amino-2-pyrazinoic acid amide using Boc2O and a base like NaH or triethylamine in a polar solvent.
- Perform halogen exchange by reacting the protected intermediate with a fluoride donor such as potassium fluoride (KF) and a phase transfer catalyst in DMSO.
- Remove the Boc protecting group using concentrated hydrochloric acid to reveal the free amino functionality.
- Convert the amino group to a hydroxyl group via diazotization using sodium nitrite in sulfuric acid at low temperatures.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers tangible strategic benefits that extend beyond mere chemical yield. The primary advantage lies in the drastic simplification of the supply chain for raw materials. By utilizing 6-bromo-3-amino-2-pyrazinoic acid amide, which is easier to source and prepare than the nitro-precursors of the past, manufacturers can reduce their dependency on specialized chemical vendors. This shift mitigates the risk of supply disruptions and provides greater leverage in price negotiations. Furthermore, the elimination of complex purification steps like column chromatography translates directly into reduced processing time and lower labor costs per kilogram of finished product.
- Cost Reduction in Manufacturing: The economic impact of this new method is profound, driven primarily by the replacement of expensive and hazardous reagents with commodity chemicals. The avoidance of phosphorus oxychloride and strictly anhydrous solvents reduces the need for specialized corrosion-resistant equipment and rigorous drying protocols. Additionally, the high yields observed in each step mean that less raw material is wasted, effectively lowering the material cost basis. The ability to use crystallization instead of chromatography for purification significantly cuts down on solvent consumption and silica gel costs, leading to substantial overall cost savings in the manufacturing budget.
- Enhanced Supply Chain Reliability: From a logistics perspective, the robustness of this process ensures consistent output quality and volume. The tolerance for mild reaction conditions means that the process is less sensitive to minor fluctuations in temperature or reagent quality, which are common variables in large-scale production. This reliability allows for more accurate forecasting and inventory planning. Moreover, the use of common solvents like acetonitrile, THF, and methanol ensures that solvent supply chains remain stable and unaffected by the volatility seen with specialty reagents. This stability is critical for maintaining continuous production schedules and meeting tight delivery deadlines for downstream API manufacturers.
- Scalability and Environmental Compliance: The environmental footprint of this synthesis is significantly smaller than that of conventional routes, aligning with increasingly strict global regulations on chemical manufacturing. The reduction in hazardous waste generation, particularly from chlorinated byproducts, simplifies waste disposal and lowers compliance costs. The process is inherently scalable, as demonstrated by the patent's emphasis on industrial applicability. The straightforward workup procedures involving filtration and extraction are easily adapted to large reactor vessels, facilitating a smooth transition from pilot plant to full commercial production without the need for extensive re-engineering of the process flow.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the practical aspects of adopting this technology.
Q: Why is the Boc protection strategy superior to direct fluorination in this synthesis?
A: Direct fluorination on the unprotected amino-pyrazine often leads to side reactions and polymerization. The Boc group stabilizes the nitrogen, allowing for cleaner nucleophilic aromatic substitution with fluoride ions, thereby significantly improving the overall yield and purity profile.
Q: What are the main drawbacks of the conventional nitro-based synthesis route?
A: Conventional routes starting from nitro precursors require harsh conditions such as refluxing in phosphorus oxychloride (POCl3) and strictly anhydrous environments. These methods typically suffer from low yields, particularly in the final hydroxylation step, and necessitate expensive column chromatography for purification.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the patented method utilizes readily available starting materials like 6-bromo-3-amino-2-pyrazinoic acid amide and avoids complex purification techniques like column chromatography. The reliance on crystallization and extraction makes it highly adaptable for multi-kilogram to ton-scale production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Fluoro-3-Hydroxy-2-Pyrazinecarboxamide Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent to production requires a partner with deep technical expertise and a commitment to quality. Our team has extensively evaluated the synthetic route described in CN102603658A and confirmed its potential for high-efficiency manufacturing. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 6-fluoro-3-hydroxy-2-pyrazinecarboxamide meets the highest industry standards for pharmaceutical intermediates.
We invite you to collaborate with us to optimize your supply chain for antiviral drug development. By leveraging our technical capabilities, you can achieve significant cost reductions and improve the reliability of your raw material sourcing. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized process can add value to your project immediately.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →