Optimizing Amlodipine Intermediate Production: Advanced Etherification Technology for Commercial Scale-Up
Optimizing Amlodipine Intermediate Production: Advanced Etherification Technology for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust synthetic routes that balance high purity with operational feasibility, particularly for critical cardiovascular medications like amlodipine. Patent CN102010361B introduces a transformative methodology for synthesizing 4-[2-(phthalimido)ethoxy]ethyl acetoacetate, a pivotal intermediate in the amlodipine value chain. This technical disclosure addresses long-standing inefficiencies in traditional etherification processes, specifically targeting the low yields and poor physical quality that have historically plagued manufacturing lines. By re-engineering the sequence of reagent addition and leveraging specific transition states, the invention offers a pathway to significantly enhance process reliability. For R&D directors and supply chain leaders, understanding this mechanistic shift is crucial for evaluating next-generation sourcing strategies. The following analysis dissects the chemical innovations within this patent and translates them into tangible commercial advantages for global pharmaceutical manufacturers seeking a reliable pharmaceutical intermediate supplier.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of this key amlodipine precursor involved the direct etherification of phthalimido ethanol with ethyl 4-chloroacetoacetate under basic conditions. In these legacy protocols, the sodium salt of phthalimido ethanol was generated first and then treated with the chloro-compound. However, this approach suffers from severe physicochemical drawbacks that hinder industrial viability. The sodium salt of the phthalimido ethanol exhibits extremely poor solubility in common organic solvents such as tetrahydrofuran (THF) or toluene, existing primarily as a suspended solid. This heterogeneity creates significant engineering challenges, including inefficient mixing, poor heat transfer, and inconsistent reaction kinetics. Furthermore, the presence of free ethyl 4-chloroacetoacetate in the reaction mixture allows for competitive side reactions where the strong base deprotonates the active methylene group rather than facilitating the desired nucleophilic substitution. This results in gas evolution, the formation of complex impurity profiles, and ultimately, a final product with suboptimal yield and appearance.
The Novel Approach
The patented innovation fundamentally alters the reaction trajectory by reversing the order of operations to prioritize the activation of the electrophile. Instead of generating the alcohol salt first, the process initiates by reacting ethyl 4-chloroacetoacetate with sodium hydride (NaH) at controlled low temperatures ranging from -10 to 5°C. This step generates a specific enolate species, referred to as Transition State A, which serves as the activated electrophilic partner. Subsequently, phthalimido ethanol is introduced into the system, where it reacts with any remaining base to form its sodium salt in situ. Because Transition State A is already present and highly reactive, the newly formed alcohol salt immediately undergoes nucleophilic substitution to form Transition State B, bypassing the accumulation of insoluble solids. This strategic reordering eliminates the stirring difficulties associated with the traditional method and prevents the degradation of the chloroacetoacetate substrate. 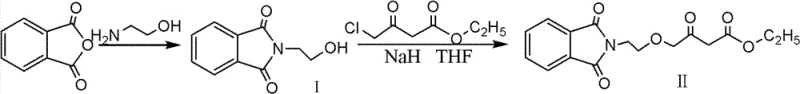
Mechanistic Insights into NaH-Mediated Etherification
The core chemical advancement lies in the kinetic control of the nucleophilic substitution through the pre-formation of the enolate. In standard Williamson ether synthesis conditions involving beta-keto esters, the acidity of the alpha-protons (active methylene) often competes with the acidity of the hydroxyl group. By pre-treating the ethyl 4-chloroacetoacetate with NaH, the system ensures that the base is consumed by the more acidic or kinetically accessible protons of the ester first, creating a stabilized anionic species. When the phthalimido ethanol is added, it reacts with residual base, but crucially, the resulting alkoxide encounters the pre-formed Transition State A immediately. This proximity effect drives the reaction towards the desired SN2 substitution pathway rather than allowing time for thermodynamic equilibration that leads to side products. The absence of free, unreacted chloroacetoacetate during the addition of the alcohol salt is the key factor that suppresses the evolution of gas and the formation of polymeric byproducts.
From an impurity control perspective, this mechanism offers a cleaner reaction profile which is essential for downstream API purification. Traditional methods often leave behind unreacted starting materials or degradation products derived from the active methylene, which can be difficult to separate due to similar polarity. The new method ensures that the limiting reagent dynamics favor the consumption of the alcohol as soon as it is deprotonated. The subsequent hydrolysis of Transition State B proceeds smoothly to yield the target 4-[2-(phthalimido)ethoxy]ethyl acetoacetate with superior physical characteristics. The patent data indicates that this mechanistic refinement translates directly into higher weight percentages and improved visual appearance of the crude product, reducing the burden on crystallization and purification units. For quality assurance teams, this implies a more consistent impurity spectrum that is easier to validate and control across multiple batches.
How to Synthesize 4-[2-(phthalimido)ethoxy]ethyl acetoacetate Efficiently
Implementing this synthesis requires precise control over temperature and stoichiometry to maximize the formation of Transition State A while minimizing side reactions. The process begins under an inert nitrogen atmosphere to prevent moisture interference with the sodium hydride. Critical parameters include maintaining the initial reaction temperature between -10 and 5°C during the addition of the chloroacetoacetate to NaH. Following the formation of the transition state, the phthalimido ethanol is added in batches to manage the exotherm and ensure complete conversion. The molar ratios are tightly defined, with a slight excess of base (NaH) relative to the alcohol to ensure full deprotonation, while the ratio of alcohol to chloroacetoacetate is kept near equimolar to optimize atom economy. Detailed standardized operating procedures for this high-efficiency synthesis route are provided in the technical guide below.
- React ethyl 4-chloroacetoacetate with sodium hydride (NaH) at low temperatures (-10 to 5°C) to generate Transition State A.
- Add phthalimido ethanol (Intermediate I) to react with remaining NaH, forming its sodium salt which immediately undergoes nucleophilic substitution with Transition State A.
- Hydrolyze the resulting Transition State B to isolate the final high-purity intermediate product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition from legacy synthesis methods to this patented approach represents a significant opportunity for cost reduction in API manufacturing. The primary economic driver is the elimination of processing bottlenecks caused by poor solubility and difficult stirring. In large-scale reactors, handling slurries of insoluble sodium salts requires specialized agitation equipment and often leads to extended batch cycles due to slow mass transfer. By shifting to a homogeneous or semi-homogeneous system where the reactive species are generated in situ, the process becomes inherently more scalable. This improved flowability reduces the risk of hot spots and runaway reactions, thereby enhancing operational safety and reducing the downtime associated with cleaning and maintenance. Consequently, manufacturers can achieve higher throughput without capital-intensive upgrades to existing infrastructure.
- Cost Reduction in Manufacturing: The avoidance of side reactions directly correlates to reduced raw material waste and lower solvent consumption for purification. In traditional processes, significant amounts of ethyl 4-chloroacetoacetate are lost to deprotonation and gas evolution, necessitating higher input loads to achieve target yields. The new method maximizes the utilization of this key starting material by channeling it exclusively into the desired etherification pathway. Furthermore, the improved quality and appearance of the crude intermediate suggest that fewer recrystallization steps or less aggressive chromatographic purification may be required. This reduction in downstream processing intensity leads to substantial cost savings in terms of solvent recovery, energy usage for distillation, and labor hours, providing a clear competitive advantage in margin-sensitive markets.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by batch failures resulting from inconsistent mixing or unpredictable reaction kinetics in heterogeneous systems. The patented method mitigates these risks by establishing a more robust and reproducible chemical environment. The ability to operate effectively at slightly elevated temperatures after the initial addition (warming to room temperature and then 45°C) allows for flexible scheduling and faster cycle times compared to processes that require cryogenic conditions throughout. This operational flexibility ensures that production schedules can be met consistently, reducing the lead time for high-purity pharmaceutical intermediates. Suppliers adopting this technology can offer more reliable delivery commitments, shielding downstream API manufacturers from volatility caused by production delays.
- Scalability and Environmental Compliance: As regulatory pressure mounts regarding waste disposal and solvent emissions, processes that minimize byproduct formation are increasingly valuable. The suppression of gas-evolving side reactions means less venting of volatile organic compounds and a simpler off-gas treatment requirement. Additionally, the higher yield per batch means that less waste sludge is generated per kilogram of product, aligning with green chemistry principles. The process is explicitly designed to be suitable for industrial production, indicating that it has been vetted for scalability beyond the laboratory bench. This readiness for commercial scale-up of complex intermediates allows companies to rapidly expand capacity to meet market demand for cardiovascular medications without encountering the typical teething problems associated with scaling poorly soluble reaction systems.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced synthesis route. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this technology into their existing supply chains.
Q: How does this new method solve the solubility issues of traditional amlodipine intermediate synthesis?
A: Traditional methods involve generating the sodium salt of phthalimido ethanol first, which has poor solubility in THF or toluene, leading to difficult stirring and heterogeneous reaction conditions. The patented method reverses the addition order, pre-forming the enolate of ethyl 4-chloroacetoacetate, ensuring a more homogeneous reaction environment and eliminating stirring bottlenecks.
Q: What specific side reactions are avoided by pre-forming Transition State A?
A: In conventional processes, the strong base can deprotonate the active methylene group of ethyl 4-chloroacetoacetate, causing gas evolution and impurity formation. By pre-reacting the chloroacetoacetate with NaH to form Transition State A before introducing the alcohol, the system avoids competitive deprotonation of the active methylene during the critical substitution phase.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the process is specifically designed for industrialization. By resolving the issues of poor solubility and difficult stirring associated with the solid sodium salt of Intermediate I, the new method ensures consistent heat transfer and mixing efficiency, which are critical parameters for safe and reliable commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-[2-(phthalimido)ethoxy]ethyl acetoacetate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the efficiency of your final API depends heavily on the quality and consistency of your starting materials. Our technical team has extensively analyzed the advancements described in patent CN102010361B and possesses the expertise to implement this optimized etherification protocol at scale. We combine deep chemical knowledge with robust manufacturing capabilities, ensuring that every batch meets stringent purity specifications. Our facilities are equipped with rigorous QC labs and advanced reactor systems capable of handling sensitive organometallic and base-mediated reactions safely. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability regardless of volume.
We invite you to collaborate with us to leverage these process improvements for your amlodipine production lines. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis that quantifies the potential efficiencies of switching to this superior synthetic route. We encourage you to contact us today to request specific COA data and route feasibility assessments tailored to your project requirements. Let us help you secure a stable, high-quality supply of critical pharmaceutical intermediates that drive your business forward.