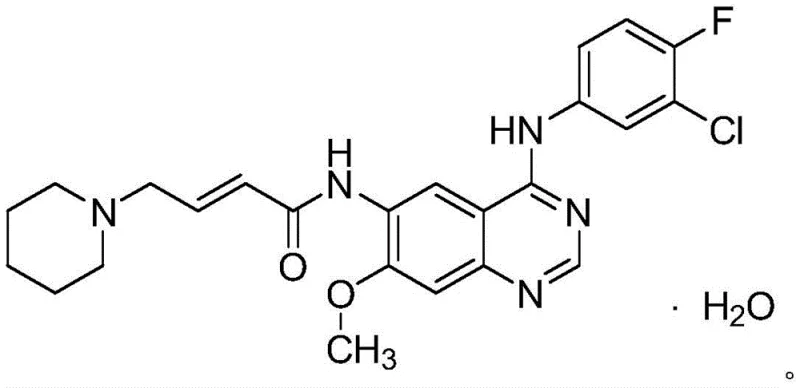
Optimizing Dacomitinib Production: Advanced Synthesis for Commercial Scale-up
Optimizing Dacomitinib Production: Advanced Synthesis for Commercial Scale-up
The pharmaceutical landscape for oncology treatments is constantly evolving, with a critical demand for efficient manufacturing processes of second-generation EGFR inhibitors like dacomitinib. Patent CN112661707B introduces a transformative preparation method that addresses longstanding bottlenecks in the synthesis of this potent kinase inhibitor. This technical disclosure outlines a robust three-step sequence involving methyl oxidation, reduction, and condensation, specifically designed to enhance overall yield and purity while simplifying operational complexity. By shifting away from noble metal catalysts and labor-intensive purification techniques, this route offers a compelling value proposition for industrial production. The structural integrity of the final monohydrate form is crucial for bioavailability, and the described method ensures consistent quality. As global demand for targeted cancer therapies rises, the ability to produce high-purity dacomitinib intermediates reliably becomes a strategic asset for supply chains. This report analyzes the technical merits and commercial implications of this patented methodology for key decision-makers in the pharmaceutical sector.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of dacomitinib has been plagued by significant technical hurdles that impede efficient commercialization. Prior art, such as the methods described in CN1972688B, often relies heavily on noble metal reduction catalysts, which introduce substantial cost burdens and supply chain vulnerabilities. The use of precious metals not only inflates raw material expenses but also necessitates rigorous and expensive removal processes to meet stringent residual metal specifications required by regulatory bodies. Furthermore, traditional routes frequently depend on column chromatography for purification, a technique that is notoriously difficult to scale beyond laboratory settings. Column chromatography consumes vast quantities of solvents, generates significant hazardous waste, and limits throughput, making it economically unviable for multi-ton production. These factors collectively result in prolonged lead times and inconsistent batch quality, creating friction for procurement managers seeking stable supply sources. The environmental footprint of such processes also poses compliance risks in an era of increasing green chemistry regulations.
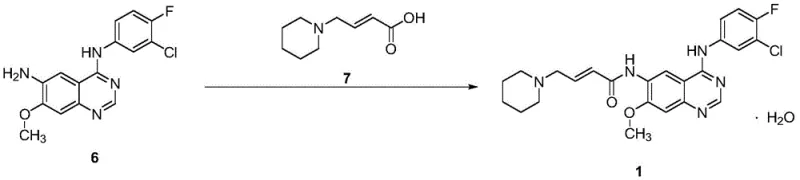
The Novel Approach
The methodology disclosed in CN112661707B represents a paradigm shift towards process intensification and cost efficiency. By replacing noble metal reduction with a sulfide-based reduction system, the new route eliminates the need for expensive catalysts and the associated purification steps. This substitution drastically simplifies the workflow, allowing for direct crystallization rather than chromatographic separation. The process utilizes readily available alkali metal hydroxides and common organic solvents, enhancing supply chain resilience. Operational simplicity is further achieved through precise temperature controls and pH adjustments that facilitate high-yield crystallization directly from the reaction mixture. This approach not only reduces the number of unit operations but also minimizes solvent consumption and waste generation. For supply chain heads, this translates to a more predictable production schedule and reduced dependency on specialized reagents. The robustness of the crystallization steps ensures that the final product meets high-purity standards consistently, addressing the critical quality concerns of R&D directors without compromising on economic feasibility.
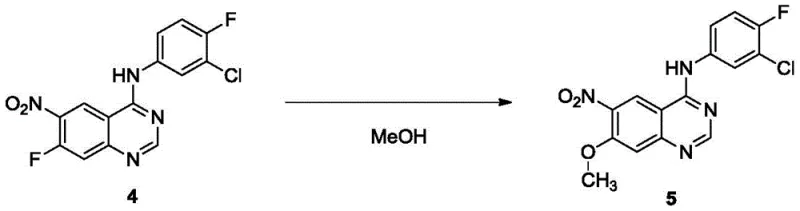
Mechanistic Insights into Methoxylation and Reduction Steps
The core of this synthetic strategy lies in the precise execution of the methoxylation and reduction transformations, which set the stage for the final coupling. The initial methoxylation step involves the reaction of a nitro-quinazoline precursor with methanol in the presence of an alkali metal hydroxide. The patent specifies a temperature range of 55°C to 65°C, which is critical for driving the nucleophilic substitution to completion while minimizing side reactions. The use of potassium hydroxide or sodium hydroxide ensures a strong basic environment necessary for the activation of methanol. Following this, the reduction of the nitro group to an amine is achieved using sodium sulfide or its hydrate in a polar aprotic solvent like N-methylpyrrolidone. This choice of reducing agent is pivotal; it selectively reduces the nitro group without affecting other sensitive functionalities on the quinazoline ring. The reaction temperature is maintained between 75°C and 85°C to optimize kinetics. The mechanism avoids the formation of azo or hydrazo byproducts often seen with other reducing agents, thereby simplifying the impurity profile. This chemical precision is essential for maintaining the integrity of the molecule throughout the synthesis.
Impurity control is inherently built into the reaction design through the selection of reagents and crystallization conditions. The use of sulfide reduction, followed by high-temperature crystallization with water addition, effectively precipitates the desired amine intermediate while leaving soluble impurities in the mother liquor. The patent details a specific water addition protocol where the mass ratio of compound to water is tightly controlled to induce crystallization at elevated temperatures, followed by cooling. This thermal cycling helps in excluding structurally similar impurities that might co-precipitate under isothermal conditions. Furthermore, the subsequent condensation step utilizes acyl chlorination with thionyl chloride, which is quenched carefully to prevent hydrolysis of the sensitive acrylamide double bond. The pH adjustment to between 8 and 9 during the final workup ensures that the product precipitates in its free base form before conversion to the salt or hydrate if necessary. These meticulous controls over reaction parameters and workup conditions result in a final product with a maximum single impurity of less than 0.25%, demonstrating superior purity compared to conventional methods.
How to Synthesize Dacomitinib Efficiently
Implementing this synthesis route requires adherence to specific operational parameters to maximize yield and safety. The process begins with the preparation of the methoxy intermediate, followed by reduction to the amine, and concludes with the amide coupling. Each step has been optimized to balance reaction rate with selectivity, ensuring that the process is robust enough for manufacturing environments. The detailed standardized synthesis steps below outline the critical control points identified in the patent data, providing a roadmap for technical teams to replicate the high yields reported. Understanding the stoichiometry and thermal profiles is essential for successful scale-up.
- Perform methoxylation of the nitro-quinazoline precursor using alkali metal hydroxide in methanol at 55-65°C.
- Execute reduction of the nitro group to an amine using sodium sulfide in an organic solvent like N-methylpyrrolidone.
- Conduct final condensation with the acrylamide side chain via acyl chlorination, followed by pH adjustment and crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthesis route offers substantial benefits for procurement and supply chain management. The elimination of noble metals and column chromatography directly translates to significant cost reduction in API manufacturing. By removing expensive catalysts and reducing solvent usage, the variable cost per kilogram of the intermediate is drastically lowered. This economic efficiency allows for more competitive pricing strategies without sacrificing margin. Additionally, the reliance on commodity chemicals like sodium sulfide and methanol enhances supply security, as these materials are widely available from multiple global vendors. This diversification reduces the risk of supply disruptions caused by single-source dependencies. For supply chain heads, the simplified process flow means shorter cycle times and higher throughput capacity. The ability to produce high-purity material without complex purification steps accelerates the time to market for downstream drug products. These factors collectively strengthen the overall resilience and cost-effectiveness of the pharmaceutical supply chain.
- Cost Reduction in Manufacturing: The removal of noble metal catalysts eliminates the need for costly scavenging resins and extensive metal testing, leading to direct savings in raw material and quality control expenses. The shift from chromatography to crystallization reduces solvent consumption by a significant margin, lowering waste disposal costs and environmental compliance burdens. Operational simplicity reduces labor hours and equipment occupancy time, further driving down the cost of goods sold. These cumulative savings create a more sustainable economic model for long-term production.
- Enhanced Supply Chain Reliability: Utilizing widely available reagents such as alkali hydroxides and sulfides mitigates the risk of raw material shortages that often plague specialized catalyst markets. The robust nature of the crystallization steps ensures consistent batch-to-batch quality, reducing the likelihood of production delays due to out-of-specification results. This reliability allows for more accurate forecasting and inventory planning, ensuring continuous availability of the intermediate for downstream formulation. A stable supply of high-quality intermediates is critical for maintaining uninterrupted drug manufacturing schedules.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, avoiding unit operations that are difficult to transfer from lab to plant. The reduction in hazardous waste generation aligns with increasingly strict environmental regulations, reducing the regulatory risk profile of the manufacturing site. Efficient solvent recovery systems can be easily integrated due to the simpler solvent profile, further enhancing the green chemistry credentials of the process. This alignment with sustainability goals is increasingly important for corporate social responsibility and regulatory approval.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this dacomitinib synthesis method. These answers are derived directly from the patent specifications and are intended to clarify the operational advantages and chemical rationale behind the process. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this route for commercial production.
Q: How does this patent improve upon previous dacomitinib synthesis methods?
A: Unlike prior art requiring noble metal reduction and column chromatography, this method utilizes sulfide reduction and crystallization, eliminating heavy metals and simplifying purification for industrial viability.
Q: What are the critical reaction conditions for the methoxylation step?
A: The methoxylation reaction requires alkali metal hydroxide (preferably KOH or NaOH) in methanol at a controlled temperature range of 55°C to 65°C to ensure high conversion and purity.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process avoids column chromatography and uses robust crystallization techniques, making it highly scalable and cost-effective for producing multi-kilogram quantities of dacomitinib intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dacomitinib Supplier
The technical potential of this synthesis route is immense, offering a clear path to efficient and high-quality production of dacomitinib intermediates. NINGBO INNO PHARMCHEM stands ready to leverage this technology, bringing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with rigorous QC labs and stringent purity specifications to ensure that every batch meets the exacting standards required for oncology drug manufacturing. We understand the critical nature of supply continuity in the pharmaceutical industry and are committed to delivering consistent quality.
We invite you to discuss how we can optimize your supply chain with this advanced manufacturing approach. Contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to support your decision-making process. Let us partner with you to secure a reliable and cost-effective source for your dacomitinib needs.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →