Advanced Synthesis of Asymmetric 4-Fluorobenzenesulfonyl-3,5-diarylmethylene-4-piperidones for Oncology Applications
Advanced Synthesis of Asymmetric 4-Fluorobenzenesulfonyl-3,5-diarylmethylene-4-piperidones for Oncology Applications
The landscape of oncology drug discovery is constantly evolving, driven by the need for agents that offer superior efficacy with reduced systemic toxicity. A pivotal advancement in this domain is documented in patent CN108191751B, which discloses a novel series of seven asymmetric 4-fluorobenzenesulfonyl substituted 3,5-diarylmethylene-4-piperidone compounds. These molecules represent a strategic evolution from traditional curcumin analogs, addressing critical limitations such as poor bioavailability and chemical instability while introducing a potent 4-fluorobenzenesulfonyl moiety that enhances metabolic stability and target binding affinity. For pharmaceutical developers seeking a reliable pharmaceutical intermediate supplier, this technology offers a robust platform for developing next-generation anti-tumor and anti-inflammatory therapeutics. The structural diversity achieved through asymmetric substitution allows for fine-tuning of physicochemical properties, ensuring that these compounds can effectively penetrate cell membranes and engage with critical apoptotic pathways.
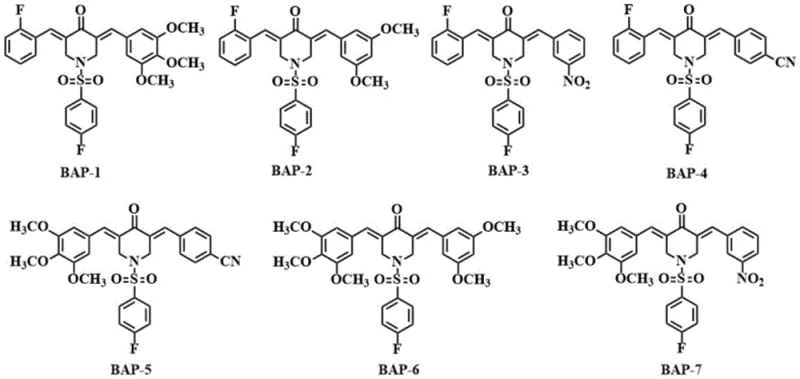
The core innovation lies in the specific arrangement of substituents on the aryl rings and the nitrogen atom. By systematically varying electron-withdrawing groups (such as fluoro, nitro, and cyano) and electron-donating groups (such as methoxy), the inventors have created a library of compounds designated BAP-1 through BAP-7. This structural modulation is not merely academic; it directly influences the molecule's polarity, solubility, and ultimately, its biological profile. The inclusion of the fluorine atom, known for its ability to enhance membrane permeability and block metabolic hotspots, combined with the sulfonamide linkage, creates a pharmacophore that is both potent and selective. This makes these intermediates highly attractive for companies focused on cost reduction in API manufacturing by minimizing late-stage failures due to poor pharmacokinetic profiles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the development of anti-cancer agents based on natural products like curcumin has been hampered by significant pharmaceutical challenges. Curcumin, while possessing broad-spectrum biological activity, exhibits extremely poor water solubility and rapid metabolic degradation in vivo, rendering it ineffective for clinical use without complex formulation strategies. Furthermore, early generations of synthetic curcumin analogs, specifically symmetric 3,5-bis(arylmethylene)-4-piperidones, often lacked the necessary selectivity between tumor and normal cells. Many of these symmetric derivatives demonstrated cytotoxicity against healthy tissues, limiting their therapeutic window. Additionally, conventional synthesis routes for piperidone derivatives often relied on harsh conditions or produced mixtures of symmetric and asymmetric products that were difficult to separate, leading to low overall yields and high purification costs. The inability to precisely control the substitution pattern on the piperidone ring meant that optimizing the structure-activity relationship (SAR) was a slow and resource-intensive process, often resulting in candidates that failed to meet the stringent requirements for high-purity pharmaceutical intermediates.
The Novel Approach
The methodology outlined in the patent data introduces a paradigm shift by focusing on asymmetric substitution and N-sulfonylation. By replacing the central diketone of curcumin with an N-substituted-4-piperidone ring, the structural stability is markedly improved. The novel approach specifically targets the nitrogen atom for modification with a 4-fluorobenzenesulfonyl group, a strategy inspired by the success of sulfonamide-containing drugs like celecoxib and glimepiride. This modification serves a dual purpose: it acts as an auxiliary binding site to enhance interaction with tumor cell targets, and it modulates the acidity and lipophilicity of the molecule to improve bioavailability. Unlike previous methods that struggled with symmetry, this process allows for the precise installation of different aryl groups at the 3 and 5 positions of the piperidone ring. This asymmetry is crucial for maximizing the coordination of the three binding sites—the 1,4-pentadienone pharmacophore, the N-substituent, and the aryl rings—thereby significantly boosting anti-tumor potency while reducing toxicity to normal cells. This targeted molecular design facilitates the commercial scale-up of complex pharmaceutical intermediates by providing a clear, reproducible pathway to high-value candidates.
Mechanistic Insights into Claisen-Schmidt Condensation and Sulfonylation
The synthesis of these advanced intermediates relies on a sophisticated yet operationally simple two-step sequence that leverages well-understood organic transformations optimized for high fidelity. The first step involves a Claisen-Schmidt condensation reaction, where 4-piperidone hydrochloride reacts with two distinct aromatic aldehydes. This reaction is catalyzed either by dry hydrogen chloride gas in acetic acid or by sodium hydroxide in methanol/water mixtures, depending on the electronic nature of the aldehyde substituents. The mechanism proceeds through the formation of an enolate or enol intermediate from the piperidone, which then undergoes nucleophilic attack on the carbonyl carbon of the aldehydes. The subsequent dehydration leads to the formation of the conjugated double bond system characteristic of the 1,4-pentadienone motif. Controlling the stoichiometry at a 1:1:1 molar ratio is critical to favoring the formation of the asymmetric product over symmetric by-products. The reaction is typically conducted at mild temperatures between 15°C and 50°C for 6 to 24 hours, ensuring that the sensitive functional groups on the aromatic rings remain intact while driving the condensation to completion.
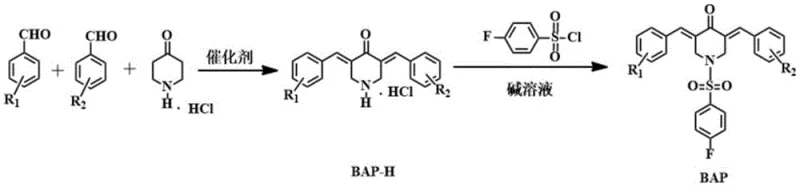
Following the condensation, the crude intermediate (BAP-H) is isolated and subjected to rigorous purification via silica gel column chromatography using a gradient of petroleum ether and ethyl acetate. This step is vital for reducing lead time for high-purity intermediates by removing unreacted starting materials and symmetric impurities before the final modification. The second step is the N-sulfonylation, where the purified intermediate reacts with 4-fluorobenzenesulfonyl chloride in a solvent such as dichloromethane or 1,2-dichloroethane. A base, typically pyridine or sodium carbonate, is employed to scavenge the hydrochloric acid generated during the reaction. This nucleophilic substitution at the nitrogen atom installs the crucial fluorobenzenesulfonyl group. The reaction proceeds smoothly at room temperature overnight, demonstrating excellent functional group tolerance. The final product is obtained after a simple workup involving acid washing to remove excess base and recrystallization or chromatography. This mechanistic clarity ensures that the process is robust and scalable, minimizing the risk of batch-to-batch variability which is a common concern in the supply of pharmaceutical intermediates.
How to Synthesize 4-Fluorobenzenesulfonyl-3,5-diarylmethylene-4-piperidones Efficiently
The preparation of these high-value compounds is designed to be accessible for industrial manufacturing while maintaining the strict quality standards required for drug development. The process begins with the precise mixing of 4-piperidone hydrochloride and two different aromatic aldehydes in a suitable solvent system, followed by the addition of the catalyst. Reaction progress is meticulously monitored using thin-layer chromatography (TLC) to determine the exact endpoint, preventing over-reaction or degradation. Once the intermediate is formed, it is isolated via suction filtration and washed with dilute sodium bicarbonate to neutralize acidity. The residue is then purified using 200-300 mesh silica gel column chromatography to yield the pure BAP-H intermediate. In the subsequent step, this intermediate is dissolved in dichloromethane and treated with 4-fluorobenzenesulfonyl chloride and a base like pyridine. After stirring overnight at room temperature, the mixture is washed with dilute hydrochloric acid and water. The final product is obtained as a yellow solid after concentration and purification. Detailed standardized synthesis steps for specific derivatives are provided in the guide below.
- Perform Claisen-Schmidt condensation between 4-piperidone hydrochloride and two distinct aromatic aldehydes in a solvent like acetic acid or methanol using a catalyst such as dry HCl gas or NaOH.
- Isolate the intermediate 3,5-diarylmethylene-N-H-4-piperidone hydrochloride (BAP-H) via filtration and purify using silica gel column chromatography.
- React the purified intermediate with 4-fluorobenzenesulfonyl chloride in dichloromethane with a base like pyridine to yield the final 4-fluorobenzenesulfonyl substituted compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition from laboratory curiosity to commercial viability is often the biggest hurdle. The synthesis route described in patent CN108191751B offers distinct logistical and economic advantages that streamline the supply chain for oncology drug development. The reliance on readily available starting materials such as 4-piperidone hydrochloride and various substituted benzaldehydes ensures a stable supply base, mitigating the risk of raw material shortages that often plague specialty chemical manufacturing. Furthermore, the reaction conditions are notably mild, operating at near-ambient temperatures and atmospheric pressure. This eliminates the need for expensive high-pressure reactors or energy-intensive heating and cooling systems, directly contributing to cost reduction in pharmaceutical intermediate manufacturing. The simplicity of the workup procedure, which primarily involves filtration and washing rather than complex distillation or extraction protocols, reduces solvent consumption and waste generation, aligning with modern green chemistry principles and environmental compliance standards.
- Cost Reduction in Manufacturing: The synthetic pathway avoids the use of precious metal catalysts or exotic reagents, relying instead on commodity chemicals like acetic acid, methanol, and pyridine. This significantly lowers the bill of materials (BOM) cost per kilogram. Additionally, the high selectivity of the reaction minimizes the formation of difficult-to-remove impurities, reducing the burden on downstream purification processes which are often the most costly part of API production. By simplifying the purification train, manufacturers can achieve substantial operational savings and faster throughput times without compromising on the purity specifications required for clinical trials.
- Enhanced Supply Chain Reliability: The robustness of the Claisen-Schmidt condensation and sulfonylation steps ensures consistent batch quality, which is critical for maintaining regulatory compliance. The process tolerances are wide enough to accommodate minor variations in raw material quality without affecting the final outcome, thereby enhancing supply chain resilience. Moreover, the scalability of the reaction from gram to multi-kilogram scales has been demonstrated through the detailed examples provided in the patent, giving procurement teams confidence in the ability to secure long-term supply agreements for clinical and commercial phases.
- Scalability and Environmental Compliance: The use of common organic solvents that are easily recovered and recycled supports sustainable manufacturing practices. The absence of heavy metals or toxic by-products simplifies waste treatment and disposal, reducing the environmental footprint of the production facility. This alignment with environmental, social, and governance (ESG) goals is increasingly important for multinational corporations selecting partners for their supply chains. The process is inherently safe, with no exothermic runaways or hazardous gas evolution, facilitating easier permitting and safer operations at large production sites.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of new chemical entities is essential for making informed sourcing decisions. The following questions address common inquiries regarding the stability, scalability, and application of these 4-fluorobenzenesulfonyl substituted piperidone derivatives. The answers are derived directly from the experimental data and technical disclosures within the patent documentation, ensuring accuracy and relevance for technical stakeholders evaluating these compounds for potential integration into their drug discovery pipelines.
Q: What represents the key structural advantage of these asymmetric piperidone derivatives over traditional curcumin analogs?
A: Unlike curcumin, which suffers from poor water solubility and instability, these asymmetric 3,5-diarylmethylene-4-piperidones feature a stable 1,4-pentadienone pharmacophore within a piperidone ring. The introduction of the 4-fluorobenzenesulfonyl group at the nitrogen position significantly enhances lipophilicity and metabolic stability while maintaining potent anti-tumor and anti-inflammatory activities.
Q: How does the synthesis route ensure high purity for pharmaceutical applications?
A: The process utilizes a controlled two-step sequence. The initial Claisen-Schmidt condensation is monitored via TLC to ensure complete conversion before isolation. Crucially, the intermediate undergoes rigorous purification using 200-300 mesh silica gel column chromatography with specific petroleum ether/ethyl acetate gradients, effectively removing unreacted aldehydes and symmetric by-products before the final sulfonylation step.
Q: Are the reaction conditions suitable for large-scale commercial manufacturing?
A: Yes, the protocol operates under mild conditions, with reaction temperatures ranging from 15°C to 50°C and reaction times between 6 to 24 hours. The use of common industrial solvents like acetic acid, methanol, and dichloromethane, combined with simple workup procedures involving filtration and washing, makes the process highly amenable to scale-up from kilogram to multi-ton production without requiring specialized high-pressure or cryogenic equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-Fluorobenzenesulfonyl-3,5-diarylmethylene-4-piperidone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a partner who can translate complex patent chemistry into reliable commercial supply. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and timeliness. We understand that the transition from bench-scale synthesis to industrial manufacturing requires more than just following a recipe; it demands a deep understanding of process safety, impurity profiling, and quality control. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, ensuring that every batch of 4-fluorobenzenesulfonyl-3,5-diarylmethylene-4-piperidone delivered meets the highest industry standards. We are committed to supporting your R&D efforts by providing high-quality intermediates that accelerate your path to clinical success.
We invite you to collaborate with us to explore the full potential of these promising anti-tumor agents. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. Whether you need small quantities for preclinical studies or large volumes for GMP manufacturing, we encourage you to contact us to request specific COA data and route feasibility assessments. Let us be your strategic partner in bringing innovative oncology therapies to market efficiently and reliably.