Revolutionizing Brimonidine Impurity Synthesis: A Safer, Scalable Commercial Route
The pharmaceutical industry faces relentless pressure to ensure the highest purity profiles for active pharmaceutical ingredients (APIs), particularly for chronic treatments like glaucoma therapy where brimonidine is a cornerstone medication. The presence of structurally related impurities, such as the debrominated analog of brimonidine, poses significant regulatory and safety challenges, necessitating robust reference standards for quality control. Patent CN112538074A introduces a transformative preparation method that addresses these critical needs by offering a concise, high-yielding synthetic pathway. This innovation not only streamlines the production of this specific impurity standard but also sets a new benchmark for efficiency in heterocyclic chemistry, moving away from hazardous legacy processes toward greener, more economically viable manufacturing protocols that align with modern Good Manufacturing Practice (GMP) expectations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of brimonidine and its related impurities has been plagued by inefficient multi-step sequences and the reliance on dangerous reagents that complicate industrial scalability. As illustrated in the reaction schemes below, traditional approaches often involve cumbersome protection-deprotection strategies or the use of highly toxic intermediates that pose severe occupational health risks. For instance, earlier literature methods utilized 4,5-dihydro-1H-imidazole-2-sulfonic acid, a reagent that is not only prohibitively expensive for large-scale procurement but also difficult to source reliably in bulk quantities. Furthermore, alternative pathways reported in patents like US6323204 necessitated the use of thiophosgene and benzene, creating a hazardous operational environment that requires extensive engineering controls and waste treatment infrastructure.
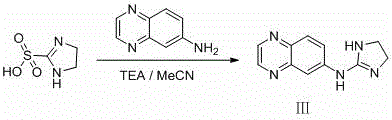
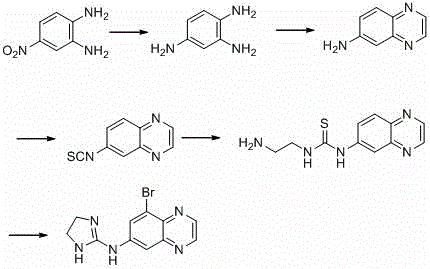
The cumulative effect of these legacy limitations is a supply chain that is fragile and cost-prohibitive. The five-step sequence required in older methods inherently accumulates yield losses at every stage, drastically reducing the overall throughput and increasing the cost of goods sold (COGS). Additionally, the handling of carcinogenic solvents like benzene and toxic gases like thiophosgene imposes strict regulatory burdens on manufacturers, often leading to production delays due to safety audits or environmental compliance issues. These factors collectively render conventional methods unsuitable for the agile, cost-sensitive demands of the contemporary generic pharmaceutical market, where speed to market and margin preservation are paramount for commercial success.
The Novel Approach
In stark contrast to these archaic methodologies, the novel approach detailed in the patent data leverages a strategic two-step synthesis that fundamentally redesigns the molecular assembly of the target impurity. By initiating the sequence with 2-(methylthio)-2-imidazoline hydroiodide, a readily available and inexpensive commodity chemical, the process immediately bypasses the sourcing bottlenecks associated with specialized sulfonic acids. The introduction of a tert-butyloxycarbonyl (Boc) protecting group in the first step serves a dual purpose: it stabilizes the reactive imidazoline nitrogen and enhances the solubility profile of the intermediate, facilitating smoother downstream processing. This strategic modification allows for a direct condensation with 6-aminoquinoxaline in the subsequent step, effectively collapsing what was once a five-step ordeal into a streamlined, high-efficiency operation.
The operational simplicity of this new route cannot be overstated, as it proceeds under mild thermal conditions using acetic acid as a benign solvent system. This shift eliminates the need for cryogenic temperatures or high-pressure reactors, significantly lowering the capital expenditure required for equipment. Moreover, the avoidance of heavy metal catalysts or toxic gaseous reagents simplifies the purification workflow, often allowing for straightforward crystallization or column chromatography without the need for complex scavenging resins. For procurement managers, this translates to a dramatic reduction in raw material volatility and a more predictable production schedule, ensuring that critical reference standards can be manufactured consistently to support the rigorous quality assurance testing required for brimonidine API batches.
Mechanistic Insights into Boc-Protection and Heterocyclic Condensation
The core chemical innovation driving this process lies in the precise manipulation of nucleophilicity and electrophilicity through the Boc-protection strategy. In the first stage, the reaction of 2-(methylthio)-2-imidazoline hydroiodide with di-tert-butyl dicarbonate (Boc2O) in the presence of a base like triethylamine creates a robust carbamate intermediate. This step is critical because it masks the secondary amine functionality, preventing unwanted polymerization or side reactions during the subsequent heating phase. The base acts to neutralize the hydroiodide salt, liberating the free base which then attacks the anhydride carbonyl of the Boc reagent. The optimization of the molar ratio of base to substrate, typically ranging from 1:1 to 1:5, ensures complete conversion while minimizing the formation of urea byproducts that could contaminate the final product.
Following protection, the mechanism shifts to a nucleophilic aromatic substitution or condensation pathway depending on the specific activation of the quinoxaline ring. When the protected imidazoline intermediate reacts with 6-aminoquinoxaline in acetic acid at temperatures between 50°C and 80°C, the amino group of the quinoxaline attacks the electrophilic carbon of the imidazoline ring. The acidic medium protonates the leaving group, facilitating the departure of the methylthio moiety and driving the equilibrium toward the formation of the desired guanidine-like linkage. This cyclization is thermodynamically favored under these conditions, and the extended reaction time of 10 to 36 hours allows for the complete consumption of starting materials, thereby maximizing the yield of the target debrominated impurity while suppressing the formation of regioisomers.
How to Synthesize Debrominated Brimonidine Efficiently
Implementing this synthesis requires careful attention to stoichiometry and temperature control to replicate the high yields reported in the patent data. The process begins with the suspension of the imidazoline salt in a chlorinated solvent, followed by the slow addition of the protecting group reagent to manage exotherms. Once the protected intermediate is isolated, it is dissolved in acetic acid along with the aminoquinoxaline partner and heated in an oil bath to ensure uniform thermal distribution. Detailed standardized operating procedures regarding workup and purification are essential to maintain the high purity levels required for analytical standards, and the full technical breakdown of these steps is provided in the guide below.
- Protect 2-(methylthio)-2-imidazoline hydroiodide with di-tert-butyl dicarbonate in the presence of triethylamine to form the Boc-intermediate.
- React the protected intermediate with 6-aminoquinoxaline in acetic acid at elevated temperatures (50-80°C) to achieve cyclization and final product formation.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain leaders and procurement officers, the adoption of this novel synthetic route represents a significant opportunity to optimize the cost structure of impurity standard production. By transitioning away from the volatile pricing of specialized sulfonic acids and the logistical nightmares of hazardous gas transport, manufacturers can achieve a much more stable and predictable cost base. The reduction in synthetic steps directly correlates to lower labor costs and reduced utility consumption, as fewer isolation and purification events are required. This efficiency gain allows for a more competitive pricing model for the final reference standard, enabling pharmaceutical companies to allocate their quality control budgets more effectively across their broader portfolio of ophthalmic products.
- Cost Reduction in Manufacturing: The elimination of expensive starting materials like 4,5-dihydro-1H-imidazole-2-sulfonic acid results in substantial raw material savings, as the new route utilizes commodity chemicals that are produced at a global scale with consistent pricing. Furthermore, the removal of toxic reagents such as thiophosgene eliminates the need for specialized scrubbing systems and hazardous waste disposal contracts, which are often significant line items in the operational budget of a chemical plant. The higher overall yield of the two-step process compared to the five-step legacy route means that less raw material is wasted per kilogram of finished product, further driving down the effective cost per gram of the impurity standard.
- Enhanced Supply Chain Reliability: Sourcing 2-(methylthio)-2-imidazoline hydroiodide is far less risky than procuring thiophosgene or benzene, as the former is a stable solid with a long shelf life and multiple global suppliers. This diversification of the supply base mitigates the risk of production stoppages due to raw material shortages or transportation restrictions on hazardous goods. Additionally, the simplified process flow reduces the lead time required to manufacture a batch, allowing for more responsive inventory management and the ability to rush orders when unexpected quality issues arise in the main API production line, ensuring continuous compliance with regulatory timelines.
- Scalability and Environmental Compliance: The use of acetic acid and dichloromethane, while requiring standard solvent recovery systems, avoids the extreme environmental liabilities associated with benzene, a known human carcinogen. This makes the process easier to permit in jurisdictions with strict environmental regulations, facilitating faster site qualification and technology transfer. The mild reaction conditions also mean that the process can be scaled from laboratory glassware to multi-ton reactors without significant re-engineering, as the heat transfer and mixing requirements remain manageable, supporting a seamless transition from R&D to commercial manufacturing without the typical scale-up pitfalls.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this technology integrates into existing quality control frameworks. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this new standard for their internal testing protocols.
Q: Why is the new synthesis route for brimonidine impurities considered superior to prior art?
A: The new route eliminates the use of highly toxic reagents like thiophosgene and benzene found in previous methods, significantly reducing safety hazards and waste disposal costs while shortening the synthetic sequence from five steps to just two.
Q: What are the key cost drivers reduced in this manufacturing process?
A: Cost reduction is achieved primarily through the use of cheap, commercially available starting materials like 2-(methylthio)-2-imidazoline hydroiodide, replacing expensive sulfonic acid derivatives, and by minimizing unit operations which lowers labor and energy consumption.
Q: Is this process suitable for large-scale GMP production?
A: Yes, the process utilizes mild reaction conditions and common solvents like acetic acid and dichloromethane, avoiding extreme pressures or temperatures, which makes it highly amenable to scale-up in standard stainless steel reactors without requiring specialized corrosion-resistant equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Debrominated Brimonidine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your final drug product depends heavily on the quality of the reference standards used to test it. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need grams for method validation or kilograms for routine QC, we can deliver with consistency. We operate stringent purity specifications and utilize rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of debrominated brimonidine meets the exacting standards required by global pharmacopeias, providing you with the confidence needed to navigate regulatory audits successfully.
We invite you to leverage our technical expertise to optimize your supply chain for ophthalmic intermediates. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the potential economic benefits of switching to this newer, safer synthesis route for your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate how our advanced manufacturing capabilities can become a strategic asset in your quest for quality excellence and operational efficiency.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →