Advanced Synthesis of Neuroprotective Intermediates: Scalable High-Purity Manufacturing Solutions
The pharmaceutical industry continuously seeks robust synthetic routes for complex neuroprotective agents, particularly those targeting Alzheimer's disease and Parkinson's syndrome. Patent CN112409201B introduces a groundbreaking preparation method for 2-hydroxy-5-[2-(4-(trifluoromethylphenyl)ethylamino)]benzoic acid, a critical intermediate in the synthesis of novel cell necrosis inhibitors. This technical disclosure addresses long-standing challenges in drug synthesis by optimizing hydroxyl protection reagents and process parameters to strictly control isomer and disubstituted impurities. The innovation lies not merely in the chemical transformation but in the strategic selection of protecting groups that enhance intermediate stability and purification efficiency. For R&D directors and procurement specialists, this patent represents a significant leap forward in achieving the stringent purity specifications required for modern API manufacturing. By fundamentally re-engineering the synthetic pathway, the method ensures that the final product quality consistently meets the rigorous demands of preparation research and clinical application standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art synthesis routes, such as those reported by Wu Yuliang, relied heavily on 2-(4-trifluoromethyl)phenethyl methanesulfonate as a key intermediate. However, this mesylate derivative suffers from inherent physical instability, characterized by a low melting point ranging between 27°C and 28°C, which makes it exceptionally difficult to purify through standard crystallization techniques. The inability to effectively refine this intermediate leads to the carryover of meta-position isomer impurities, specifically 2-(3-trifluoromethyl)phenethyl methanesulfonate, into subsequent reaction steps. Furthermore, the condensation reaction using this unstable intermediate tends to generate high levels of disubstituted impurities, such as 2-hydroxy-5-[N,N-bis(2-(4-(trifluoromethyl)phenyl)ethyl)amino]methyl benzoate. These impurities persist through hydrolysis and contaminate the final active pharmaceutical ingredient, posing severe risks to patient safety and regulatory compliance. Consequently, the conventional route is increasingly viewed as inadequate for meeting the escalating quality control standards of the global pharmaceutical market.
The Novel Approach
The patented method overcomes these deficiencies by substituting the methanesulfonate group with more robust protecting groups such as p-toluenesulfonyl, benzenesulfonyl, or acetyl groups. This strategic modification significantly improves the physical properties of the intermediate, allowing for effective purification via crystallization using solvents like n-heptane or ethyl acetate. By removing meta-isomer impurities at the intermediate stage, the process prevents their propagation into the final product, thereby drastically enhancing overall purity. Additionally, the new protecting groups modulate the reactivity during the condensation step, effectively suppressing the formation of disubstituted byproducts that plagued previous methods. The result is a synthesis route that delivers the target benzoic acid derivative with exceptional purity levels exceeding 99.9% and significantly improved yields. This approach not only solves the technical bottlenecks of the past but also establishes a reliable foundation for cost reduction in pharmaceutical intermediate manufacturing by minimizing waste and rework.

Mechanistic Insights into Hydroxyl Protection and Condensation
The core of this synthetic innovation lies in the initial protection step where 2-(4-trifluoromethyl)phenethyl alcohol reacts with a sulfonyl chloride reagent to form a stable sulfonate ester. The selection of the protecting group R is critical, with p-toluenesulfonyl chloride emerging as the preferred reagent due to its optimal balance of reactivity and steric bulk. This reaction is typically conducted in organic solvents like toluene at controlled temperatures between 0°C and 30°C to prevent side reactions. The resulting sulfonate ester exhibits superior crystallinity compared to its mesylate counterpart, enabling the removal of trace isomeric impurities through a dedicated crystallization step performed between 20°C and 30°C. This purification is essential because it ensures that only the correct para-isomer proceeds to the coupling stage, fundamentally altering the impurity profile of the entire synthesis. The mechanistic precision here ensures that the downstream condensation reaction starts with a high-purity substrate, which is a prerequisite for achieving the final API quality targets.
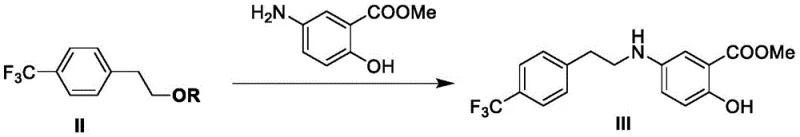
Following the protection and purification, the intermediate undergoes a nucleophilic substitution reaction with methyl 5-aminosalicylate to form the key ester intermediate. This condensation is carried out in the presence of a base such as triethylamine at temperatures ranging from 80°C to 90°C. Strict temperature control is vital here, as excessive heat can promote the formation of disubstituted impurities where two amine molecules react with a single sulfonate. The patent describes a novel salt formation step where the crude condensation product is converted into a hemisulfate salt using sulfuric acid. This salt formation induces crystallization, allowing for the separation of the desired product from unreacted starting materials and organic byproducts. Finally, the ester group is hydrolyzed under acidic conditions with nitrogen bubbling to yield the target carboxylic acid. This final hydrolysis step is optimized to ensure complete conversion while maintaining the integrity of the sensitive amine linkage, resulting in a final product with negligible levels of critical impurities.

How to Synthesize 2-hydroxy-5-[2-(4-(trifluoromethylphenyl)ethylamino)]benzoic acid Efficiently
Implementing this synthesis route requires careful attention to the specific process parameters outlined in the patent to ensure reproducibility and high quality. The procedure begins with the protection of the starting alcohol, followed by a critical crystallization purification that sets the stage for high purity. The subsequent condensation and hydrolysis steps must be monitored closely, particularly regarding temperature and acid concentration, to avoid degradation or side reactions. Detailed standardized synthesis steps are provided below to guide technical teams in replicating this high-efficiency process.
- Protect 2-(4-trifluoromethyl)phenethyl alcohol using p-toluenesulfonyl chloride to form a stable sulfonate intermediate.
- Condense the protected intermediate with methyl 5-aminosalicylate in toluene with triethylamine to form the ester derivative.
- Hydrolyze the ester derivative using sulfuric acid under nitrogen bubbling to obtain the final high-purity benzoic acid target.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers substantial advantages for procurement managers and supply chain heads looking to optimize their sourcing strategies for complex pharmaceutical intermediates. The elimination of unstable intermediates reduces the risk of batch failures and production delays, ensuring a more consistent supply of high-quality materials. By simplifying the purification process through effective crystallization, the method reduces the reliance on expensive chromatographic separations, leading to significant cost savings in manufacturing. Furthermore, the use of common industrial solvents and reagents enhances the scalability of the process, making it suitable for large-volume production without requiring specialized equipment. These factors collectively contribute to a more resilient supply chain capable of meeting the demanding timelines of drug development projects.
- Cost Reduction in Manufacturing: The switch to stable sulfonate intermediates eliminates the need for complex and costly purification steps associated with low-melting mesylates. By enabling purification through simple crystallization, the process reduces solvent consumption and energy usage significantly. The higher yields achieved through impurity control mean less raw material is wasted, directly lowering the cost of goods sold. Additionally, the reduction in disubstituted impurities minimizes the need for reprocessing or discarding off-spec batches, further enhancing economic efficiency.
- Enhanced Supply Chain Reliability: The robustness of the new intermediates ensures that production can proceed without the interruptions caused by unstable materials degrading during storage or transport. This stability allows for larger batch sizes and longer inventory holding times without quality degradation. Consequently, suppliers can offer more reliable lead times and maintain consistent stock levels to meet sudden demand surges. The use of widely available reagents also mitigates the risk of supply chain disruptions caused by the scarcity of specialized chemicals.
- Scalability and Environmental Compliance: The process is designed with commercial scale-up in mind, utilizing standard reaction conditions that are easily transferable from pilot plant to full-scale production. The ability to remove impurities via crystallization reduces the generation of hazardous waste streams associated with chromatographic purification. This aligns with increasingly strict environmental regulations and reduces the cost of waste disposal. The overall efficiency of the route supports sustainable manufacturing practices while maintaining high productivity levels.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the patent data to provide accurate guidance for stakeholders evaluating this technology. Understanding these details is crucial for making informed decisions about process adoption and supplier selection.
Q: How does the new protecting group strategy improve impurity profiles?
A: By replacing the unstable mesylate group with a tosyl or besyl group, the intermediate gains higher melting points and better crystallinity, allowing for the effective removal of meta-isomer impurities before the condensation step.
Q: What are the critical parameters for controlling disubstituted impurities?
A: Maintaining the condensation reaction temperature between 80°C and 90°C and utilizing specific solvent systems like toluene are critical to minimizing the formation of disubstituted byproducts during the amine coupling.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process utilizes common solvents like toluene and standard reagents, and the intermediates can be purified via crystallization, making it highly scalable for industrial manufacturing from 100 kgs to 100 MT.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-hydroxy-5-[2-(4-(trifluoromethylphenyl)ethylamino)]benzoic acid Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis, leveraging deep technical expertise to bring complex pathways like this to commercial reality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest international standards. Our commitment to quality ensures that the technical advantages of this patent are fully realized in the final product delivered to your facility.
We invite you to collaborate with us to optimize your supply chain and reduce costs through advanced manufacturing techniques. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate our capability to support your project goals. Let us be your partner in delivering high-quality pharmaceutical intermediates efficiently.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →