Advanced Manufacturing of 4-Hydroxy Aurone Compounds for Pharmaceutical Applications
Advanced Manufacturing of 4-Hydroxy Aurone Compounds for Pharmaceutical Applications
The pharmaceutical industry continuously seeks efficient pathways to access bioactive scaffolds, and the synthesis of 4-hydroxy aurone compounds represents a critical area of development due to their potent biological profiles. Patent CN102993142A discloses a robust preparation method that addresses longstanding challenges in constructing the aurone core with high regioselectivity and yield. These compounds are renowned for their diverse pharmacological activities, including anti-tumor properties, cell proliferation inhibition, and potential applications in managing diabetic syndromes. The disclosed technology leverages a strategic protection-deprotection sequence that significantly streamlines the manufacturing process compared to legacy techniques. By utilizing 2',6'-dihydroxyacetophenone as a key starting material, the method ensures precise control over the substitution pattern, which is vital for maintaining biological efficacy. This technical breakthrough provides a solid foundation for the commercial scale-up of complex pharmaceutical intermediates, offering a reliable supply chain solution for drug developers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of aurone derivatives has been plagued by cumbersome reaction sequences and harsh operating conditions that hinder efficient production. Prior art, such as the method described in patent CN101914081, relies on the reaction of substituted phenols with chloroacetonitrile using zinc chloride catalysts in diethyl ether. This conventional approach necessitates extremely low temperatures ranging from minus ten to ten degrees Celsius for extended periods of up to seventy-two hours, followed by high-temperature hydrolysis steps. Such drastic thermal swings not only consume significant energy but also complicate process control and safety management in a plant setting. Furthermore, the multi-step nature of these older routes often leads to cumulative yield losses and the generation of difficult-to-remove impurities. The lack of explicit disclosure for preparing specific 4-hydroxy substituted variants in previous literature further limits the utility of these methods for modern drug discovery programs requiring precise structural analogs.
The Novel Approach
In stark contrast, the novel methodology presented in CN102993142A introduces a streamlined four-step sequence that operates under much milder and more controllable conditions. The innovation lies in the initial selective protection of one hydroxyl group on the 2',6'-dihydroxyacetophenone backbone using chloromethyl methyl ether, which directs subsequent reactions to the desired position. This strategic modification allows for a smooth condensation with benzaldehyde derivatives followed by an efficient cyclization step catalyzed by mercury acetate. The reaction conditions are notably gentle, with key steps proceeding at moderate temperatures between forty-five and fifty-five degrees Celsius or under reflux in common solvents like ethanol and pyridine. This reduction in thermal stress and reaction complexity translates directly into shorter processing times and simplified operational procedures. The ability to achieve high conversion rates without the need for cryogenic cooling or extended reaction durations marks a significant advancement in the manufacturing of these valuable fine chemicals.
Mechanistic Insights into Regioselective Cyclization and Protection
The core of this synthetic success lies in the meticulous management of regiochemistry through the use of a methoxymethyl (MOM) protecting group. In the first stage, 2',6'-dihydroxyacetophenone reacts with chloromethyl methyl ether in the presence of anhydrous potassium carbonate in acetone. This step selectively masks one of the phenolic hydroxyls, preventing unwanted side reactions during the subsequent aldol condensation. The resulting intermediate, 2'-hydroxy-6'-methoxymethoxyacetophenone, retains a free hydroxyl group ortho to the acetyl moiety, which is crucial for the final ring closure. Following this, the protected ketone undergoes a base-catalyzed Claisen-Schmidt condensation with various 4-R-benzaldehydes. Using pyrrolidine or sodium hydroxide in ethanol facilitates the formation of the chalcone intermediate with high stereoselectivity for the trans-alkene geometry. This precise control over the molecular architecture ensures that the subsequent cyclization occurs exclusively to form the five-membered furanone ring characteristic of aurones, rather than alternative isomers.
The cyclization step itself is a masterpiece of oxidative heterocycle formation, utilizing mercury(II) acetate as a Lewis acid catalyst in pyridine. This reagent promotes the intramolecular attack of the phenolic oxygen onto the alpha-beta unsaturated ketone system, closing the ring to form the aurone core. The reaction is typically conducted at temperatures between 115 and 125 degrees Celsius, providing sufficient energy to overcome the activation barrier for ring closure while maintaining stability of the sensitive functional groups. Following cyclization, the final step involves the removal of the MOM protecting group under acidic conditions using hydrochloric acid in methanol. This deprotection reveals the free 4-hydroxyl group, completing the synthesis of the target molecule. The entire pathway is designed to minimize byproduct formation, ensuring that the final crude product requires minimal purification, which is a critical factor for cost-effective manufacturing.
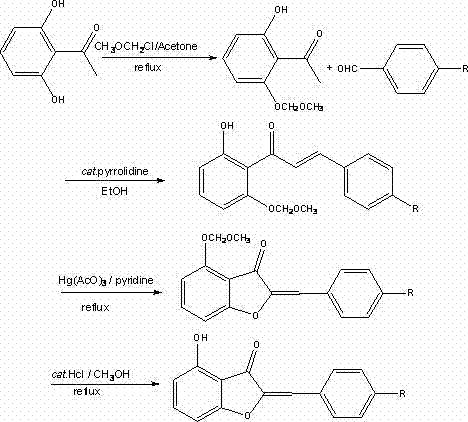
How to Synthesize 4-Hydroxy Aurone Efficiently
Implementing this synthesis protocol requires careful attention to solvent quality and stoichiometric ratios to maximize the efficiency of each transformation. The process begins with the protection step in anhydrous acetone, where moisture control is essential to prevent hydrolysis of the chloromethyl methyl ether reagent. Subsequent condensation and cyclization steps rely on the purity of the intermediates, suggesting that intermediate isolation or rigorous in-process monitoring via TLC is advisable. The final deprotection is straightforward but requires precise pH control to ensure complete removal of the protecting group without degrading the aurone scaffold. For detailed operational parameters, including specific molar ratios, stirring speeds, and workup procedures, please refer to the standardized guide below which outlines the critical process parameters derived from the patent examples.
- Protect one hydroxyl group of 2',6'-dihydroxyacetophenone using chloromethyl methyl ether in acetone with potassium carbonate.
- Perform aldol condensation between the protected intermediate and 4-R-benzaldehyde using pyrrolidine in ethanol.
- Cyclize the chalcone intermediate using mercury(II) acetate in pyridine at elevated temperatures.
- Remove the methoxymethyl protecting group using hydrochloric acid in methanol to yield the final 4-hydroxy aurone.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this synthetic route offers compelling economic benefits driven primarily by the accessibility and low cost of the starting materials. The use of 2',6'-dihydroxyacetophenone and substituted benzaldehydes leverages commodity chemicals that are widely available in the global market, reducing the risk of supply bottlenecks. Unlike methods requiring exotic catalysts or specialized reagents that may have long lead times, the reagents here—such as potassium carbonate, pyrrolidine, and mercury acetate—are standard industrial chemicals. This availability ensures a stable supply chain and mitigates the volatility often associated with niche pharmaceutical intermediates. Furthermore, the high yields reported in the patent examples indicate a material-efficient process, meaning less raw material is wasted per kilogram of final product. This efficiency directly correlates to lower variable costs and a reduced environmental footprint regarding waste disposal.
- Cost Reduction in Manufacturing: The elimination of extreme temperature requirements, such as cryogenic cooling, significantly lowers energy consumption and capital expenditure on specialized refrigeration equipment. The simplified workflow reduces labor hours and reactor occupancy time, allowing for higher throughput in existing facilities. By avoiding the multi-step degradation and low yields associated with older zinc-catalyzed methods, the overall cost of goods sold is substantially optimized. The use of common solvents like acetone, ethanol, and methanol further simplifies solvent recovery and recycling processes, contributing to additional operational savings.
- Enhanced Supply Chain Reliability: The reliance on robust, well-understood chemical transformations enhances the predictability of production schedules. Since the reaction conditions are mild and tolerant to minor variations, the risk of batch failure is minimized, ensuring consistent delivery timelines to downstream customers. The versatility of the method to accommodate various R-groups on the benzaldehyde ring means that a single production line can be adapted to manufacture a family of related derivatives. This flexibility allows suppliers to respond quickly to changing market demands without the need for extensive retooling or new process validation campaigns.
- Scalability and Environmental Compliance: While the use of mercury salts requires careful handling, the defined workup procedures involving filtration and acidic quenching allow for effective containment and disposal in compliance with environmental regulations. The process generates fewer organic byproducts compared to traditional routes, simplifying wastewater treatment requirements. The high atom economy of the condensation and cyclization steps ensures that the majority of the input mass ends up in the desired product. This alignment with green chemistry principles facilitates easier regulatory approval and supports the sustainability goals of modern pharmaceutical companies seeking responsible partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of 4-hydroxy aurone derivatives using this patented methodology. These insights are derived directly from the experimental data and process descriptions found in the source intellectual property. Understanding these nuances is essential for R&D teams evaluating the feasibility of this route for their specific pipeline candidates. The answers provide clarity on yield expectations, impurity profiles, and the adaptability of the process to different substituents.
Q: What are the primary advantages of this synthesis method over conventional routes?
A: This method utilizes a regioselective protection strategy that simplifies the reaction pathway compared to older methods requiring extreme temperature variations and prolonged reaction times. It offers higher overall yields and uses readily available starting materials like 2',6'-dihydroxyacetophenone.
Q: Is the use of mercury catalysts a concern for large-scale production?
A: While mercury(II) acetate is used for the cyclization step, the process includes specific workup procedures involving acidic treatment and filtration to isolate the product. Industrial scale-up would require appropriate heavy metal scavenging protocols to meet stringent pharmaceutical purity specifications.
Q: Can this method accommodate various substituents on the benzaldehyde ring?
A: Yes, the synthetic route is highly versatile and tolerates various substituents (R groups) on the benzaldehyde component, such as bromo or dimethylamino groups, allowing for the generation of a diverse library of aurone derivatives for biological screening.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-Hydroxy Aurone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in accelerating drug development timelines. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial plant is seamless. We are committed to meeting stringent purity specifications through our rigorous QC labs, which utilize advanced analytical techniques to verify the identity and quality of every batch. Our expertise in handling complex organic syntheses, including those involving sensitive functional groups and cyclization reactions, positions us as an ideal partner for your aurone derivative requirements.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits of switching to this more efficient manufacturing method. We encourage potential partners to contact us for specific COA data and route feasibility assessments to ensure that our capabilities align perfectly with your supply chain objectives. Let us collaborate to bring your next-generation therapeutic candidates to market faster and more cost-effectively.