Advanced Purification Technology for Methotrexate Intermediates Ensuring Commercial Scalability
Advanced Purification Technology for Methotrexate Intermediates Ensuring Commercial Scalability
The pharmaceutical industry constantly faces the challenge of controlling trace impurities in complex oncology drugs, particularly when regulatory limits are stringent. A recent technological breakthrough detailed in patent CN114249731A introduces a sophisticated refining method for methotrexate intermediates that directly addresses the persistence of "Stubborn Impurity C." This impurity, which must be maintained below 0.5% according to European Pharmacopoeia EP9.0 standards, has historically been difficult to remove using conventional purification techniques. The disclosed innovation utilizes a specific salt formation strategy involving p-toluenesulfonic acid to selectively crystallize the desired intermediate while leaving impurity precursors in the mother liquor. For global procurement teams and R&D directors, this represents a critical advancement in ensuring batch-to-batch consistency and regulatory compliance for antifolate therapeutics used in treating tumors and rheumatoid arthritis.
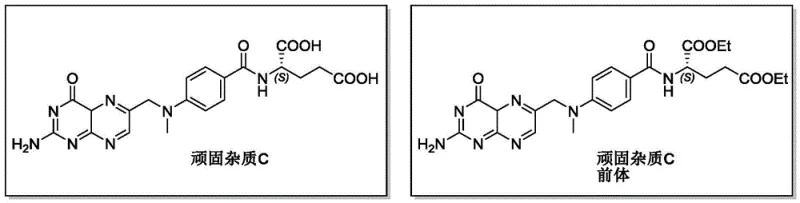
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis routes for methotrexate, such as those described in prior art like WO2012/074496a1, often involve converting raw materials like 4-methylamino benzoyl zinc glutamate into esters before coupling with pteridine derivatives. While these methods can produce the target molecule, they frequently struggle with the accumulation of specific degradation products or side-reaction byproducts known as Stubborn Impurity C. As illustrated in the structural analysis, this impurity shares a close structural homology with the target intermediate, making separation via standard chromatography or simple recrystallization inefficient and costly. Furthermore, conventional post-treatment methods often fail to reduce the precursor of this impurity below the critical 0.5% threshold required for final API release, leading to significant batch rejection rates and supply chain disruptions for manufacturers relying on older synthetic pathways.
The Novel Approach
The innovative approach presented in the patent data fundamentally shifts the purification paradigm by targeting the intermediate stage rather than the final API. By reacting the crude intermediate Int1 with p-toluenesulfonic acid monohydrate, the process forms a distinct crystalline salt (Formula I) that exhibits different solubility characteristics compared to the impurity precursor. This selective crystallization effectively purifies the intermediate before the final hydrolysis step. Experimental results demonstrate that this salt formation technique can reduce the precursor content of Stubborn Impurity C from approximately 0.89% in crude material to as low as 0.24% in the purified salt. This proactive purification strategy ensures that subsequent hydrolysis steps yield a final methotrexate product that comfortably meets international pharmacopoeia standards without requiring additional, expensive downstream processing.
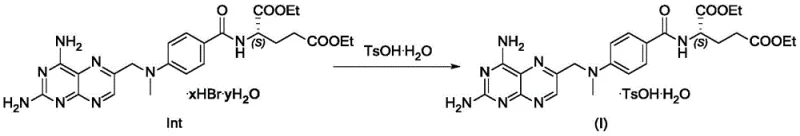
Mechanistic Insights into TsOH-Mediated Salt Formation Purification
The core mechanism driving this purification success lies in the differential solubility and lattice energy of the p-toluenesulfonate salt of the methotrexate intermediate. When p-toluenesulfonic acid is introduced to the reaction system containing Int1, it protonates the basic nitrogen centers of the pteridine ring or interacts with the amine functionalities to form a stable ionic lattice. This specific salt form, designated as Formula I, precipitates out of the solution under controlled solvent conditions, while the structurally similar impurity precursor remains solvated in the mother liquor due to its inability to fit into the growing crystal lattice of the desired salt. The process is highly sensitive to solvent polarity; data indicates that protic solvents like ethanol facilitate this selective precipitation better than aprotic solvents like DMF, likely due to hydrogen bonding networks that stabilize the crystal growth of the target salt while keeping impurities dissolved.
Furthermore, the control of water content during the crystallization phase is a critical mechanistic parameter. The addition of water acts as an anti-solvent, reducing the solubility of the target salt and driving the equilibrium towards precipitation. However, the ratio of water to organic solvent must be precisely managed; too little water results in poor yield as the salt remains in solution, while excessive water can co-precipitate impurities or lead to oiling out, which compromises purity. The patent data highlights that a water-to-ethanol volume ratio between 1.5:1 and 3.5:1 optimizes this balance, achieving high recovery rates while maintaining the rigorous impurity profile necessary for pharmaceutical grade intermediates. This mechanistic understanding allows for robust process control during scale-up.
How to Synthesize Methotrexate Intermediate Formula I Efficiently
The synthesis of the purified intermediate involves a straightforward yet precise sequence of dissolution, reaction, and controlled crystallization. Operators must first ensure the complete dissolution of p-toluenesulfonic acid in the chosen alcohol solvent before introducing the crude intermediate to prevent localized acidity spikes that could degrade the sensitive pteridine ring. Following the addition of the intermediate, the system requires a period of equilibration to ensure homogeneous salt formation before the anti-solvent is introduced. The detailed standardized synthetic steps for this high-efficiency purification process are outlined in the guide below.
- Dissolve p-toluenesulfonic acid monohydrate in anhydrous ethanol or methanol at room temperature with stirring.
- Add the crude intermediate Int1 to the acid solution and stir until completely dissolved.
- Dropwise add purified water to the system to induce crystallization, then filter and dry to obtain the purified Formula I salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this refining technology offers substantial strategic benefits beyond mere technical compliance. By integrating this purification step early in the synthesis tree, manufacturers can significantly mitigate the risk of batch failures at the final API stage, which are exponentially more costly in terms of lost time and materials. The ability to consistently produce intermediates with impurity levels well below regulatory limits enhances supply chain reliability, ensuring that downstream formulation partners receive materials that require minimal quality assurance intervention. This stability is crucial for maintaining continuous production schedules for essential oncology medications where market demand is inelastic and interruptions can have severe clinical consequences.
- Cost Reduction in Manufacturing: The implementation of this salt formation process eliminates the need for complex and expensive chromatographic separations or multiple recrystallization cycles that were previously necessary to tackle Stubborn Impurity C. By utilizing commodity chemicals like p-toluenesulfonic acid and ethanol, the direct material costs remain low while the overall process efficiency is enhanced through higher effective yields of usable intermediate. This streamlined approach reduces the consumption of high-boiling polar aprotic solvents like DMA, which are not only costly but also require energy-intensive recovery systems, thereby lowering the overall operational expenditure associated with solvent management and waste disposal.
- Enhanced Supply Chain Reliability: The robustness of this method against variations in crude input quality provides a buffer against supply chain volatility. Since the purification step is highly effective at scrubbing out precursors even from batches of Int1 with slightly elevated impurity profiles, suppliers can maintain consistent output quality despite minor fluctuations in upstream raw materials. This resilience reduces the lead time for high-purity pharmaceutical intermediates by minimizing the need for re-processing or quarantine periods, allowing for faster turnover and more predictable delivery schedules to global clients who depend on just-in-time inventory models for their drug manufacturing operations.
- Scalability and Environmental Compliance: From an environmental and safety perspective, the shift towards ethanol-water solvent systems represents a significant improvement over processes relying heavily on chlorinated solvents or toxic amides. Ethanol is a green solvent with a favorable safety profile, simplifying the regulatory burden associated with worker exposure and environmental discharge. The mild reaction conditions, operating effectively at ambient temperatures between 20°C and 30°C, further reduce the energy footprint of the manufacturing process. This alignment with green chemistry principles not only facilitates easier permitting for commercial scale-up but also appeals to increasingly eco-conscious stakeholders in the pharmaceutical value chain who prioritize sustainable manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this refining technology. These insights are derived directly from the experimental data and comparative examples provided in the patent literature, offering a clear picture of the process capabilities and limitations. Understanding these details is essential for technical teams evaluating the feasibility of integrating this method into existing production lines.
Q: How does this refining method impact the levels of Stubborn Impurity C?
A: The method significantly reduces the precursor of Stubborn Impurity C in the intermediate stage. By converting Int1 into the p-toluenesulfonate salt (Formula I), the precursor content is lowered to below 0.5%, ensuring the final methotrexate API meets the strict European Pharmacopoeia limit of ≤0.5%.
Q: What are the optimal solvent conditions for this purification process?
A: Experimental data indicates that lower alcohols such as ethanol and methanol, or tetrahydrofuran, provide superior results compared to polar aprotic solvents like DMF or DMA. Specifically, an ethanol-water system with a volume ratio favoring water addition for crystallization yields the highest purity and lowest impurity profiles.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process operates under mild conditions (20-30°C) and utilizes common, scalable solvents like ethanol and water. The crystallization step is robust, and the overall yield remains high (around 77% in optimized examples), making it highly viable for industrial scale-up without requiring exotic reagents or extreme temperatures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Methotrexate Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust manufacturing infrastructure. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated purification techniques described in recent patents can be seamlessly translated into industrial practice. We maintain stringent purity specifications across all our product lines, supported by rigorous QC labs equipped with advanced analytical instrumentation to verify that every batch of methotrexate intermediate meets the exacting standards required for oncology drug synthesis.
We invite you to collaborate with us to optimize your supply chain for antifolate therapeutics. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. By leveraging our refined manufacturing processes, you can secure a stable source of high-quality intermediates that minimize downstream processing risks. Please contact us today to request specific COA data and route feasibility assessments that demonstrate how our capabilities align with your long-term production goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →