Advanced Synthesis of 6,6-Dimethyl-3-Azabicyclo Hexane for Commercial Pharmaceutical Manufacturing
The pharmaceutical industry continuously seeks robust synthetic pathways for complex intermediates that drive the production of life-saving medications. Patent CN114989067B introduces a groundbreaking method for synthesizing 6,6-dimethyl-3-azabicyclo [3.1.0] hexane, a critical building block in the manufacturing of Paritaprevir and other Hepatitis C protease inhibitors. This technical insight report analyzes the proprietary chemistry disclosed in the patent, highlighting its potential to revolutionize supply chain stability for global pharmaceutical manufacturers. The disclosed route offers a distinct advantage over traditional methods by eliminating hazardous reagents and significantly streamlining the operational workflow. By leveraging a novel sequence of ring-opening, functionalization, and cyclization reactions, this method ensures high purity and exceptional yield consistency. For R&D directors and procurement specialists, understanding the nuances of this synthesis is vital for securing a reliable supply of high-purity pharmaceutical intermediates. The following analysis dissects the chemical logic, operational safety, and commercial viability of this innovative process.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 6,6-dimethyl-3-azabicyclo [3.1.0] hexane has relied on precursors such as methyl chrysanthemate or ethyl chrysanthemate, which are primarily associated with pesticide manufacturing and carry prohibitive costs for pharmaceutical applications. These conventional routes often necessitate the use of ethyl diazoacetate, a small molecular azide compound that presents severe safety hazards, including a high risk of explosion if not handled with extreme caution. Furthermore, the traditional synthesis of caronic anhydride, a key intermediate in these old pathways, generates substantial environmental burdens, with reports indicating the production of 40 to 50 tons of high-salt wastewater for every single ton of anhydride synthesized. This excessive waste generation not only complicates regulatory compliance but also inflates the overall cost of goods sold due to expensive waste treatment requirements. The multi-step nature of these legacy processes often results in cumulative yield losses, making them economically unviable for large-scale commercial production of high-value API intermediates.
The Novel Approach
In stark contrast, the method disclosed in patent CN114989067B utilizes 6,6-dimethyl-3-oxabicyclo [3.1.0] hexane-2-ketone as a starting material, which is more readily available and significantly safer to handle. This innovative route bypasses the need for dangerous diazo compounds entirely, replacing them with stable amino group sources and sulfonylating agents that are standard in fine chemical manufacturing. The process is characterized by its operational simplicity, requiring only three main synthetic steps to reach the target structure, thereby reducing the time and resources needed for production. By avoiding the generation of high-salt wastewater and hazardous byproducts, this method aligns with modern green chemistry principles and environmental regulations. The strategic design of the reaction sequence ensures that the core bicyclic structure is preserved and functionalized efficiently, leading to a much cleaner reaction profile. This shift represents a substantial technological leap, offering a reliable pharmaceutical intermediate supplier pathway that prioritizes both safety and economic efficiency.
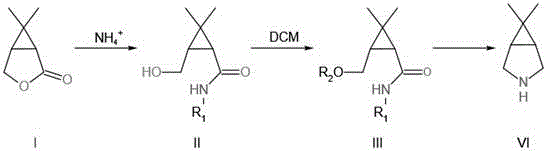
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this synthetic breakthrough lies in the precise manipulation of the bicyclic ring system through a sequence of nucleophilic attacks and reductive cyclizations. The first step involves the reaction of the starting oxabicyclo ketone with an amino group compound, such as benzylamine or aqueous ammonia, under controlled thermal conditions. This nucleophilic attack opens the oxygen-containing ring, establishing the nitrogen backbone essential for the final azabicyclo structure while maintaining the integrity of the three-membered carbocycle. Subsequent sulfonylation of the resulting hydroxyl group activates the molecule for intramolecular cyclization, creating a leaving group that facilitates the closure of the nitrogen-containing ring. The use of alkaline conditions during this stage ensures that the reaction proceeds smoothly without compromising the stereochemistry of the sensitive bicyclic framework. This careful orchestration of functional group transformations is critical for achieving the high purity required for pharmaceutical applications.
The final stage of the synthesis employs a sophisticated reduction strategy using sodium borohydride and boron trifluoride, followed by catalytic hydrogenation. This dual-reduction approach effectively converts the cyclic amide intermediate into the desired amine, removing the carbonyl oxygen and any protecting groups simultaneously. The patent details two distinct methods for this final transformation, offering flexibility depending on the specific substituents present on the nitrogen atom. Method 1 prioritizes ring closure before reduction, while Method 2 reverses this order, allowing chemists to optimize conditions based on substrate solubility and reactivity. Both pathways converge on the same high-yield target, demonstrating the robustness of the chemical design. The ability to fine-tune these final steps ensures that impurity profiles remain minimal, a key concern for R&D directors overseeing process validation and regulatory filings.
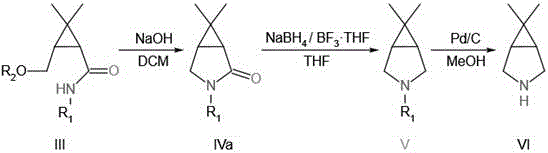
How to Synthesize 6,6-Dimethyl-3-Azabicyclo [3.1.0] Hexane Efficiently
The synthesis of this complex pharmaceutical intermediate requires precise control over reaction parameters to ensure optimal yield and safety. The patented procedure outlines a standardized protocol that begins with the careful addition of amino sources to the ketone substrate under a nitrogen atmosphere to prevent oxidation. Following the initial ring opening, the reaction mixture undergoes rigorous workup procedures, including extraction and drying, to isolate the amide intermediate with high purity. The subsequent sulfonylation step must be performed at low temperatures to control exothermicity, followed by a controlled warm-up to drive the reaction to completion. These operational details are critical for scaling the process from laboratory benchtop to industrial reactor volumes. For a comprehensive guide on the specific molar ratios, temperature profiles, and workup techniques, please refer to the standardized synthesis steps provided below.
- React 6,6-dimethyl-3-oxabicyclo [3.1.0] hexane-2-ketone with an amino group compound to form the open-ring amide intermediate.
- Treat the amide intermediate with a sulfonyl group compound under alkaline conditions to activate the hydroxyl group for cyclization.
- Perform ring-closing reaction followed by boron-mediated reduction and hydrogenolysis to yield the final azabicyclo target compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers profound advantages for procurement managers and supply chain heads looking to optimize costs and mitigate risks. The elimination of hazardous ethyl diazoacetate removes a significant safety liability from the manufacturing process, reducing insurance costs and the need for specialized explosion-proof infrastructure. Furthermore, the reduction in waste generation translates directly into lower environmental compliance costs and simpler waste disposal logistics. The high yields reported in the patent examples suggest that raw material utilization is maximized, which is a key driver for cost reduction in manufacturing. By shortening the synthetic sequence to just three main steps, the overall production lead time is drastically simplified, allowing for faster response to market demand fluctuations. These factors combine to create a supply chain that is not only more cost-effective but also more resilient to disruptions.
- Cost Reduction in Manufacturing: The replacement of expensive pesticide-grade starting materials with dedicated pharmaceutical intermediates significantly lowers the raw material cost base. Additionally, the avoidance of heavy metal catalysts or complex purification steps reduces the operational expenditure associated with catalyst recovery and product polishing. The high efficiency of the reaction steps means less solvent and energy are consumed per kilogram of product, contributing to substantial cost savings. This economic efficiency makes the process highly competitive for large-scale commercial production of complex pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The use of stable, commercially available reagents ensures that the supply chain is not dependent on niche or hazardous materials that may face regulatory restrictions. The robustness of the reaction conditions allows for consistent production quality, reducing the risk of batch failures that can disrupt supply continuity. This reliability is crucial for maintaining the production schedules of downstream API manufacturers who depend on a steady flow of high-quality intermediates. The process design inherently supports a stable and predictable supply chain.
- Scalability and Environmental Compliance: The process is designed with industrial scale-up in mind, utilizing standard unit operations such as extraction, distillation, and filtration that are easily implemented in existing manufacturing facilities. The significant reduction in hazardous waste generation simplifies environmental permitting and reduces the long-term liability associated with waste storage and treatment. This alignment with green chemistry principles enhances the sustainability profile of the manufacturing process, appealing to environmentally conscious stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic route. These answers are derived directly from the technical disclosures and experimental data provided in the patent documentation. They are intended to clarify the operational benefits and safety profile of the method for potential manufacturing partners. Understanding these details is essential for making informed decisions about process adoption and supply chain integration.
Q: Why is this synthetic route safer than conventional chrysanthemic acid methods?
A: Conventional methods often utilize ethyl diazoacetate, which poses significant explosion risks and generates high volumes of hazardous waste. This patented route avoids dangerous azide compounds entirely, utilizing stable ketone starting materials and standard reduction conditions.
Q: What are the yield advantages of this method for industrial scale-up?
A: The patent data demonstrates step yields consistently above 90%, with the final hydrogenolysis step achieving up to 98% yield. This high efficiency minimizes raw material loss and reduces the burden on downstream purification processes.
Q: Can this process be adapted for large-scale commercial production?
A: Yes, the process is designed for industrial applicability, utilizing common solvents like dichloromethane and tetrahydrofuran, and avoiding extreme pressure or temperature conditions that complicate reactor design and safety protocols.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6,6-Dimethyl-3-Azabicyclo [3.1.0] Hexane Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of this intermediate in the global supply chain for Hepatitis C treatments. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required by top-tier pharmaceutical companies. We are committed to delivering high-purity pharmaceutical intermediates that adhere to the highest quality standards, supporting your R&D and commercial manufacturing goals. Our technical team is ready to assist in the seamless transfer of this patented technology to our production lines.
We invite you to contact our technical procurement team to discuss how we can support your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of adopting this synthetic route for your supply chain. We encourage you to reach out for specific COA data and route feasibility assessments to ensure this process aligns with your manufacturing capabilities. Let us partner with you to optimize your production of complex pharmaceutical intermediates and drive your project forward.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →