Scalable Synthesis of 5-Aminobenzo[b][1,8]naphthyridine Antitumor Intermediates via TfOH Catalysis
The pharmaceutical industry is constantly seeking robust and scalable pathways to access novel heterocyclic scaffolds with potent biological activity. Patent CN110305128B, published in August 2021, introduces a significant advancement in the preparation of 5-aminobenzo[b][1,8]naphthyridine compounds, a class of molecules exhibiting promising antitumor properties. This invention details a streamlined synthetic route that utilizes trifluoromethanesulfonic acid (TfOH) as a highly efficient catalyst for intramolecular cyclization. Unlike previous methodologies that relied on harsh Lewis acids or concentrated sulfuric acid at extreme temperatures, this novel approach operates under mild conditions, offering superior yields and broader substrate compatibility. For R&D teams and procurement specialists alike, this represents a critical opportunity to secure a reliable pharmaceutical intermediate supplier capable of delivering high-purity building blocks for oncology drug discovery programs.
![General chemical structure of 5-aminobenzo[b][1,8]naphthyridine derivatives showing variable R and R' substituents](/insights/img/5-aminobenzo-naphthyridine-synthesis-pharma-supplier-20260305202653-01.png)
The core innovation lies in the ability to diversify the molecular structure at multiple positions (R and R') while maintaining a consistent and high-yielding cyclization step. The resulting compounds have demonstrated inhibitory activity against several human cancer cell lines, including nasopharyngeal carcinoma, liver cancer, and bladder cancer. By shifting the paradigm from thermal-intensive processes to acid-catalyzed cyclization at near-ambient temperatures, this technology not only improves chemical efficiency but also aligns with modern green chemistry principles by reducing energy consumption and simplifying downstream processing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
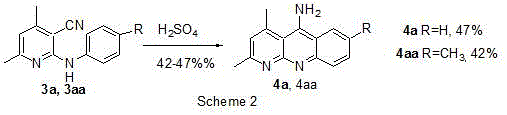
Prior to this invention, the synthesis of benzo[b][1,8]naphthyridine derivatives was fraught with significant operational challenges and inefficiencies. Historical literature, such as the work by Afflooeaei et al. in 2000, described a cyclization reaction catalyzed by aluminum trichloride (AlCl3) requiring temperatures as high as 200°C. As illustrated in the reaction scheme below, this thermal stress often led to decomposition of sensitive functional groups and resulted in mediocre yields, typically around 59% for the parent compound 4a. Furthermore, the use of stoichiometric amounts of Lewis acids like AlCl3 generates substantial amounts of aluminum waste, complicating the purification process and increasing the environmental burden of the manufacturing operation.

Another conventional route reported by Shramm et al. utilized concentrated sulfuric acid (H2SO4) as the cyclization promoter. While this avoided the extreme temperatures of the AlCl3 method, it still suffered from limited scope and moderate yields ranging between 42% and 47%. The corrosive nature of concentrated sulfuric acid poses safety risks during scale-up and requires specialized equipment resistant to strong acid corrosion. Additionally, the harsh acidic environment can lead to side reactions such as sulfonation of the aromatic rings or hydrolysis of the nitrile group, thereby generating difficult-to-remove impurities that compromise the purity profile required for pharmaceutical applications.
The Novel Approach
The methodology disclosed in CN110305128B fundamentally transforms this landscape by employing trifluoromethanesulfonic acid (TfOH) in 1,2-dichloroethane (DCE) as the reaction medium. This superacid catalyst enables the intramolecular cyclization to proceed efficiently at temperatures ranging from room temperature to merely 60°C. The dramatic reduction in thermal energy input not only preserves the integrity of thermally labile substituents but also significantly enhances the reaction yield, with many examples achieving yields above 80% and some reaching as high as 91%. This shift allows for the synthesis of a much wider array of derivatives, including those with electron-donating and electron-withdrawing groups that would otherwise degrade under traditional conditions.
Mechanistic Insights into TfOH-Catalyzed Intramolecular Cyclization
The success of this synthetic strategy hinges on the unique activation mechanism provided by trifluoromethanesulfonic acid. In the final cyclization step, the precursor molecule, a 4,6-disubstituted-2-N-aryl-3-nitrile pyridine derivative, undergoes an electrophilic aromatic substitution-like process. The strong acidity of TfOH likely protonates the nitrogen of the nitrile group or activates the adjacent carbon center, making it highly susceptible to nucleophilic attack by the electron-rich aromatic ring attached at the 2-position. This intramolecular attack closes the third ring of the naphthyridine system, forming the stable benzo[b][1,8]naphthyridine core. The use of DCE as a solvent is crucial here, as it provides a non-nucleophilic, polar environment that stabilizes the cationic intermediates without interfering with the reaction pathway.
From an impurity control perspective, the mildness of the TfOH protocol is paramount. In the older AlCl3 and H2SO4 methods, the aggressive reaction conditions often promoted polymerization of the aniline precursors or hydrolysis of the nitrile functionality into amides or carboxylic acids. By contrast, the TfOH-catalyzed reaction proceeds rapidly at low temperatures, kinetically favoring the desired cyclization over these degradation pathways. This results in a cleaner crude reaction mixture, which simplifies the subsequent purification steps. The patent describes a straightforward workup involving neutralization with saturated sodium bicarbonate, filtration, and standard column chromatography, indicating that the impurity profile is manageable and suitable for further processing into active pharmaceutical ingredients (APIs).
How to Synthesize 5-Aminobenzo[b][1,8]naphthyridine Efficiently
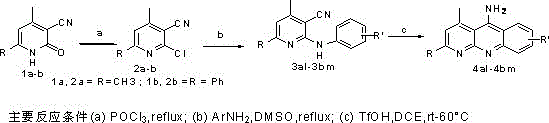
The synthesis is executed in a logical three-step sequence that begins with commercially available pyridone derivatives. First, the hydroxyl group is activated via chlorination using phosphorus oxychloride (POCl3) under reflux to generate a reactive 2-chloro intermediate. Second, this chloro-species undergoes nucleophilic aromatic substitution with a variety of substituted anilines in dimethyl sulfoxide (DMSO) at 110-130°C to install the necessary aryl amine moiety. Finally, the key cyclization is performed using the TfOH/DCE system. Detailed standardized operating procedures for each stage, including specific molar ratios, temperature ramps, and quenching protocols, are outlined in the comprehensive guide below.
- Step 1: React 4,6-disubstituted-2-oxo-1,2-dihydropyridine-3-carbonitrile with POCl3 under reflux to generate the 2-chloro intermediate.
- Step 2: Perform nucleophilic substitution of the chloro-intermediate with various substituted anilines in DMSO at 110-130°C to form the N-aryl precursor.
- Step 3: Execute intramolecular cyclization of the N-aryl precursor using trifluoromethanesulfonic acid (TfOH) in 1,2-dichloroethane at room temperature to 60°C.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this TfOH-catalyzed route offers tangible strategic benefits beyond mere chemical elegance. The transition from high-temperature batch processes to mild, solution-phase chemistry directly translates into reduced operational expenditures. By eliminating the need for specialized high-temperature reactors and the extensive energy input required to maintain 200°C conditions, manufacturers can achieve significant cost reduction in API intermediate manufacturing. Furthermore, the avoidance of stoichiometric metal salts like aluminum chloride removes the costly and time-consuming heavy metal clearance steps typically required before a compound can advance to clinical trials, thereby streamlining the regulatory filing process.
- Cost Reduction in Manufacturing: The new process drastically lowers energy consumption by operating at near-ambient temperatures compared to the legacy 200°C methods. Additionally, the use of catalytic amounts of TfOH rather than stoichiometric Lewis acids reduces raw material costs and minimizes waste disposal fees associated with hazardous metal sludge. The higher yields observed (often exceeding 80%) mean less starting material is wasted, directly improving the overall cost-of-goods sold (COGS) for the final intermediate.
- Enhanced Supply Chain Reliability: The starting materials for this route, including substituted pyridones and various anilines, are commodity chemicals available from multiple global suppliers, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions ensures consistent batch-to-batch reproducibility, which is critical for maintaining long-term supply contracts with pharmaceutical partners. The simplified workup procedure also shortens the production cycle time, allowing for faster turnaround on custom synthesis orders.
- Scalability and Environmental Compliance: Scaling this process from gram to kilogram quantities is straightforward due to the liquid-phase nature of all reaction steps and the absence of extreme pressure requirements. The use of DCE and DMSO, while requiring proper handling, is well-established in industrial settings with existing recovery and recycling infrastructure. This facilitates compliance with increasingly stringent environmental regulations regarding volatile organic compounds (VOCs) and hazardous waste generation, positioning the manufacturer as a sustainable partner in the pharmaceutical value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these naphthyridine derivatives. The answers are derived directly from the experimental data and specifications provided in the patent documentation, ensuring accuracy for technical decision-makers evaluating this technology for their pipeline.
Q: What are the advantages of using TfOH over traditional Lewis acids like AlCl3 for this synthesis?
A: The patent demonstrates that trifluoromethanesulfonic acid (TfOH) allows for cyclization at much milder temperatures (room temperature to 60°C) compared to the harsh 200°C required for AlCl3 catalysis. This results in significantly higher yields (up to 91% vs 59%) and better tolerance for diverse functional groups on the aromatic ring.
Q: Does this method support large-scale production of these intermediates?
A: Yes, the process utilizes standard organic solvents like 1,2-dichloroethane and DMSO, and avoids extremely high-pressure or cryogenic conditions. The workup involves simple filtration and column chromatography, which are readily adaptable for kilogram-to-ton scale manufacturing in a GMP environment.
Q: What biological activity do these compounds exhibit?
A: According to the patent data, the synthesized 5-aminobenzo[b][1,8]naphthyridine derivatives show potent in vitro antitumor activity against human nasopharyngeal carcinoma (CNE-2), liver cancer (HepG-2), gastric cancer (MGC-803), and bladder cancer (T-24) cell lines, with IC50 values often in the low micromolar range.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Aminobenzo[b][1,8]naphthyridine Supplier
As the demand for novel antitumor agents continues to rise, securing a supply of high-quality heterocyclic intermediates is essential for accelerating drug discovery timelines. NINGBO INNO PHARMCHEM leverages deep expertise in organic synthesis to master complex pathways like the TfOH-catalyzed cyclization described in CN110305128B. Our facility is equipped with extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements whether you are in the early lead optimization phase or preparing for clinical supply. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 5-aminobenzo[b][1,8]naphthyridine meets the exacting standards required for pharmaceutical development.
We invite you to collaborate with us to optimize your supply chain for these critical oncology intermediates. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data for our catalog compounds and conduct detailed route feasibility assessments for your custom targets, ensuring a seamless transition from bench-scale research to commercial manufacturing.