Scalable Synthesis of Potent CDK8 Inhibitors for Oncology Drug Development
Scalable Synthesis of Potent CDK8 Inhibitors for Oncology Drug Development
The landscape of oncology drug discovery is rapidly evolving towards highly selective kinase inhibitors that minimize off-target toxicity. A pivotal advancement in this domain is detailed in Chinese Patent CN115991705A, which discloses a novel series of 3-(1H-pyrrolo[2,3-b]pyridin-5-yl)benzoyl derivatives. These compounds have demonstrated remarkable potency as Cyclin-Dependent Kinase 8 (CDK8) inhibitors, offering a promising therapeutic avenue for treating aggressive malignancies such as acute myeloid leukemia, colorectal cancer, and melanoma. The patent outlines a robust chemical framework where the core scaffold is meticulously engineered to maximize binding affinity while maintaining favorable pharmacokinetic properties. For pharmaceutical developers and procurement specialists, understanding the synthetic accessibility and structural versatility of these molecules is critical for securing a reliable supply chain for next-generation cancer therapies.
![General chemical structure of 3-(1H-pyrrolo[2,3-b]pyridin-5-yl)benzoyl derivatives showing variable R1 and R2 substituents](/insights/img/pyrrolo-pyridine-benzoyl-derivatives-cdk8-inhibitor-supplier-20260306062608-01.png)
Beyond the biological efficacy, the true value proposition of this technology lies in its manufacturing feasibility. The disclosed preparation methods utilize well-established transition-metal catalyzed cross-coupling reactions, specifically the Suzuki-Miyaura protocol, which is renowned for its tolerance to functional groups and scalability. By leveraging a modular synthetic strategy, the patent enables the rapid generation of a diverse library of analogs through the variation of R1 and R2 substituents. This flexibility allows medicinal chemists to fine-tune physicochemical properties such as solubility and metabolic stability without overhauling the entire production process. Consequently, this represents a significant opportunity for cost reduction in pharmaceutical intermediate manufacturing, as the route avoids exotic reagents and extreme reaction conditions that typically drive up operational expenses.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the functionalization of fused heterocyclic systems like pyrrolo[2,3-b]pyridines has presented substantial challenges in process chemistry. Conventional approaches often suffer from poor regioselectivity, where electrophilic substitution occurs at multiple positions on the electron-rich pyrrole ring, leading to complex mixtures of isomers that are difficult and costly to separate. Furthermore, direct coupling on the unprotected nitrogen atom can lead to catalyst poisoning or undesired N-alkylation side products, drastically reducing overall yield. Traditional methods also frequently rely on harsh acidic or basic conditions that can degrade sensitive functional groups on the benzoyl moiety, limiting the scope of accessible derivatives. These inefficiencies create bottlenecks in the supply chain, resulting in long lead times for high-purity intermediates and inconsistent batch-to-batch quality that complicates regulatory filings.
The Novel Approach
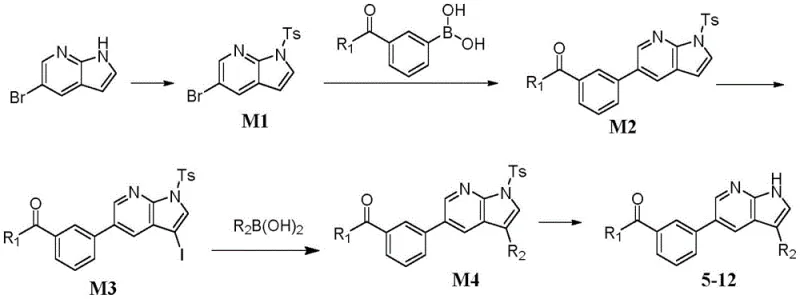
The methodology presented in the patent overcomes these hurdles through a strategic protection-deprotection sequence combined with modern palladium catalysis. By initially protecting the pyrrole nitrogen with a tosyl (Ts) group, the synthesis effectively masks the reactive site, directing subsequent halogenation exclusively to the desired C3 position of the pyrrole ring. This regiocontrol is paramount for ensuring the structural integrity of the final CDK8 inhibitor. Following the installation of the halogen handle, a second Suzuki coupling introduces diverse aryl or heteroaryl groups with high fidelity. The final deprotection step cleanly reveals the active NH moiety under mild basic conditions. This streamlined approach not only simplifies purification but also significantly enhances the reproducibility of the synthesis, making it ideally suited for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Pd-Catalyzed Suzuki-Miyaura Coupling
The core transformation driving the assembly of this molecular scaffold is the palladium-catalyzed Suzuki-Miyaura cross-coupling reaction. Mechanistically, this cycle begins with the oxidative addition of the aryl halide (either the bromo-azaindole derivative or the halogenated intermediate) to the zero-valent palladium catalyst, forming an organopalladium(II) species. This step is facilitated by the use of robust ligand systems such as dppf (1,1'-bis(diphenylphosphino)ferrocene), which stabilizes the metal center and accelerates the reaction kinetics. Subsequently, the organoboron reagent, activated by a base like potassium carbonate or sodium hydroxide, undergoes transmetallation with the palladium complex. This transfers the organic fragment to the metal center, setting the stage for the final reductive elimination step that forms the new carbon-carbon bond and regenerates the active catalyst. The efficiency of this cycle is evidenced by the high yields reported in the examples, often exceeding 75% even on multi-gram scales.
Impurity control is another critical aspect addressed by the mechanistic design of this route. The use of specific bases and solvent systems, such as the dioxane/water mixture described in Example 1, optimizes the solubility of inorganic byproducts while maintaining the stability of the organic intermediates. The protection of the nitrogen atom prevents the formation of N-linked byproducts, which are common impurities in azaindole chemistry. Additionally, the choice of mild deprotection conditions ensures that the sensitive amide or aldehyde functionalities on the benzoyl ring remain intact. This high level of chemoselectivity results in crude products with purity levels often surpassing 95%, thereby reducing the burden on downstream chromatographic purification and lowering the overall cost of goods sold for the final active pharmaceutical ingredient.
How to Synthesize 3-(1H-pyrrolo[2,3-b]pyridin-5-yl)benzoyl Derivatives Efficiently
The synthesis of these high-value CDK8 inhibitor intermediates follows a logical, step-wise progression that balances reactivity with selectivity. The process initiates with the preparation of the key heterocyclic building block, followed by sequential coupling events to construct the biaryl backbone. Each step has been optimized to minimize waste and maximize throughput, utilizing reagents that are readily sourced from global chemical suppliers. The protocol emphasizes the importance of rigorous temperature control and inert atmosphere techniques to maintain catalyst activity throughout the reaction course. For process chemists looking to implement this route, the following standardized steps provide a foundational guide for laboratory and pilot-scale production.
- Perform Suzuki-Miyaura coupling between bromo-substituted benzoyl precursors and pyrrolo[2,3-b]pyridine boronic esters using Pd catalysts.
- Execute selective halogenation (chlorination or iodination) on the pyrrole ring to introduce functional handles for further diversification.
- Conduct a second Suzuki coupling with diverse aryl/heteroaryl boronic acids followed by deprotection to yield the final high-purity derivatives.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic benefits that extend beyond simple molecule acquisition. The reliance on commodity chemicals such as bromo-benzaldehydes, boronic acids, and standard palladium catalysts means that the supply chain is resilient to disruptions often associated with bespoke or proprietary reagents. The modularity of the synthesis allows for flexible inventory management, where a single protected intermediate can be diverted to produce multiple final analogs based on real-time demand. This agility reduces the risk of obsolescence and ensures that capital is not tied up in slow-moving specialized stock. Furthermore, the high purity achieved directly from the reactor minimizes the need for extensive recrystallization or preparative HPLC, leading to substantial cost savings in manufacturing overhead.
- Cost Reduction in Manufacturing: The elimination of complex separation processes for regioisomers significantly lowers processing time and solvent consumption. By achieving high selectivity through the Ts-protection strategy, the process avoids the yield losses typically associated with purifying difficult isomeric mixtures. This efficiency translates directly into a lower cost per kilogram of the final intermediate, allowing for more competitive pricing in the generic and innovative drug markets. Additionally, the use of aqueous workups and standard extraction techniques simplifies waste treatment, further reducing environmental compliance costs.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, including 5-bromo-7-azaindole and various substituted phenylboronic acids, are produced by multiple vendors globally, mitigating the risk of single-source dependency. The robustness of the Suzuki coupling reaction ensures consistent output even when scaling from grams to kilograms, providing supply chain heads with confidence in delivery schedules. This reliability is crucial for maintaining continuous clinical trial material supply and supporting eventual commercial launch timelines without interruption.
- Scalability and Environmental Compliance: The reaction conditions employed, such as moderate temperatures (40°C to 80°C) and atmospheric pressure, are inherently safer and easier to scale than high-pressure hydrogenations or cryogenic reactions. The solvent systems used are largely recyclable, aligning with green chemistry principles and reducing the environmental footprint of the manufacturing process. This alignment with sustainability goals not only meets regulatory expectations but also enhances the corporate social responsibility profile of the pharmaceutical products derived from this technology.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these CDK8 inhibitor intermediates. The answers are derived directly from the experimental data and structural analysis provided in the patent documentation, ensuring accuracy and relevance for technical decision-makers. Understanding these details helps stakeholders evaluate the feasibility of integrating these compounds into their existing drug development pipelines.
Q: What is the primary therapeutic target of these pyrrolo[2,3-b]pyridine derivatives?
A: These compounds are designed as potent inhibitors of Cyclin-Dependent Kinase 8 (CDK8), showing significant efficacy against acute myeloid leukemia and other solid tumors.
Q: How does the Ts-protection strategy improve the synthesis yield?
A: The tosyl (Ts) protection group on the pyrrole nitrogen prevents unwanted side reactions during the halogenation and cross-coupling steps, ensuring high regioselectivity and purity.
Q: Are the starting materials for this synthesis commercially available?
A: Yes, the key building blocks such as 5-bromo-7-azaindole and various substituted phenylboronic acids are widely available commodity chemicals, facilitating easy scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-(1H-pyrrolo[2,3-b]pyridin-5-yl)benzoyl Derivatives Supplier
As the demand for targeted oncology therapies continues to surge, having a partner capable of delivering complex heterocyclic intermediates with precision is essential. NINGBO INNO PHARMCHEM stands at the forefront of this capability, leveraging extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of palladium-catalyzed reactions, including efficient metal scavenging to meet stringent purity specifications. With rigorous QC labs and a commitment to quality assurance, we ensure that every batch of 3-(1H-pyrrolo[2,3-b]pyridin-5-yl)benzoyl derivatives meets the highest international standards for clinical and commercial use.
We invite pharmaceutical companies and research institutions to collaborate with us to accelerate their CDK8 inhibitor programs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline constraints. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you secure a stable, high-quality supply of these critical pharmaceutical intermediates to drive your innovation forward.