Scalable Synthesis of Novel Terephthalamide Derivatives for Advanced Antiviral Drug Development
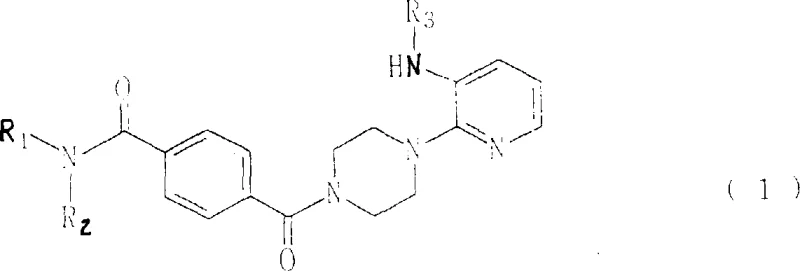
The pharmaceutical landscape for antiviral therapeutics is undergoing a significant paradigm shift, driven by the urgent need for non-nucleoside inhibitors that offer improved safety profiles over traditional therapies. Patent CN1254334A introduces a groundbreaking class of novel terephthalamide derivatives, specifically designed to combat the proliferation of critical viruses such as Hepatitis B (HBV) and Human Immunodeficiency Virus (HIV). Unlike conventional nucleoside analogs which often suffer from high toxicity and drug resistance, these new chemical entities operate through a distinct mechanism targeting the reverse transcription process. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediate suppliers, understanding the structural versatility and synthetic accessibility of these compounds is paramount. The patent details a robust framework where substituents R1, R2, and R3 can be extensively modified to optimize pharmacokinetic properties, offering a fertile ground for developing next-generation antiviral agents with enhanced efficacy and reduced side effects.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the development of anti-HBV and anti-HIV medications has been dominated by nucleoside reverse transcriptase inhibitors (NRTIs). While effective, these compounds present substantial challenges in both clinical application and chemical manufacturing. Clinically, they are frequently associated with severe mitochondrial toxicity and the rapid emergence of resistant viral strains, necessitating constant structural modifications. From a manufacturing perspective, the synthesis of nucleoside analogs is notoriously complex, often requiring multi-step protection and deprotection strategies on sugar moieties, chiral resolutions, and the use of hazardous phosphorylating agents. These factors contribute to exorbitant production costs and supply chain vulnerabilities. Furthermore, the purification of nucleoside intermediates often involves difficult chromatographic separations to remove closely related impurities, creating bottlenecks in cost reduction in API manufacturing initiatives.
The Novel Approach
The technology disclosed in CN1254334A offers a transformative alternative by utilizing a terephthalamide scaffold. This non-nucleoside approach bypasses the metabolic liabilities of sugar-based drugs while maintaining potent inhibitory activity against viral polymerases. The core innovation lies in the modular connectivity of the molecule, linking a substituted pyridyl-piperazine moiety to a terephthalic acid derivative via stable amide bonds. This structural design not only enhances metabolic stability but also simplifies the synthetic pathway significantly. By employing standard amidation and nucleophilic substitution reactions, manufacturers can achieve high yields under relatively mild conditions. This shift represents a strategic advantage for commercial scale-up of complex pharmaceutical intermediates, as it leverages well-established industrial chemistry rather than specialized nucleoside synthesis capabilities, thereby ensuring greater supply continuity and lower entry barriers for production.
Mechanistic Insights into Dual-Target Viral Inhibition
The biological efficacy of these terephthalamide derivatives stems from their ability to disrupt the viral replication cycle at two critical junctures: the reverse transcriptase (RT) activity and the RNaseH domain. The reverse transcription step is essential for converting viral RNA into DNA, a prerequisite for integration into the host genome. The specific arrangement of the pyridyl-piperazine group allows the molecule to bind effectively to the RT enzyme, sterically hindering its function. Moreover, the patent data indicates that certain derivatives exhibit remarkable inhibition of the RNaseH activity, an often-overlooked target that is crucial for removing the RNA template from the RNA-DNA hybrid. This dual-inhibition mechanism reduces the likelihood of resistance development compared to single-target agents. The structural flexibility provided by the R1 and R2 groups on the amide nitrogen allows for fine-tuning of the molecule's lipophilicity and hydrogen bonding capacity, optimizing its fit within the enzyme's active site. This precise molecular engineering ensures that the compounds maintain high potency while minimizing off-target interactions with human cellular machinery.
From a chemical stability and impurity control perspective, the amide linkage in the terephthalamide core provides exceptional robustness against hydrolysis under physiological conditions, ensuring the drug reaches its target intact. The synthesis strategy described allows for the introduction of diverse functional groups such as hydroxyalkyl, alkoxyalkyl, and heterocyclic amines, which can be strategically placed to improve solubility and bioavailability. For quality control teams, the predictable nature of the amidation reaction means that impurity profiles are generally cleaner and easier to characterize compared to the complex byproduct mixtures seen in nucleoside chemistry. This clarity in the impurity spectrum facilitates faster regulatory approval processes and more consistent batch-to-batch quality, which is a critical metric for any high-purity pharmaceutical intermediate intended for clinical use.
How to Synthesize Terephthalamide Derivatives Efficiently
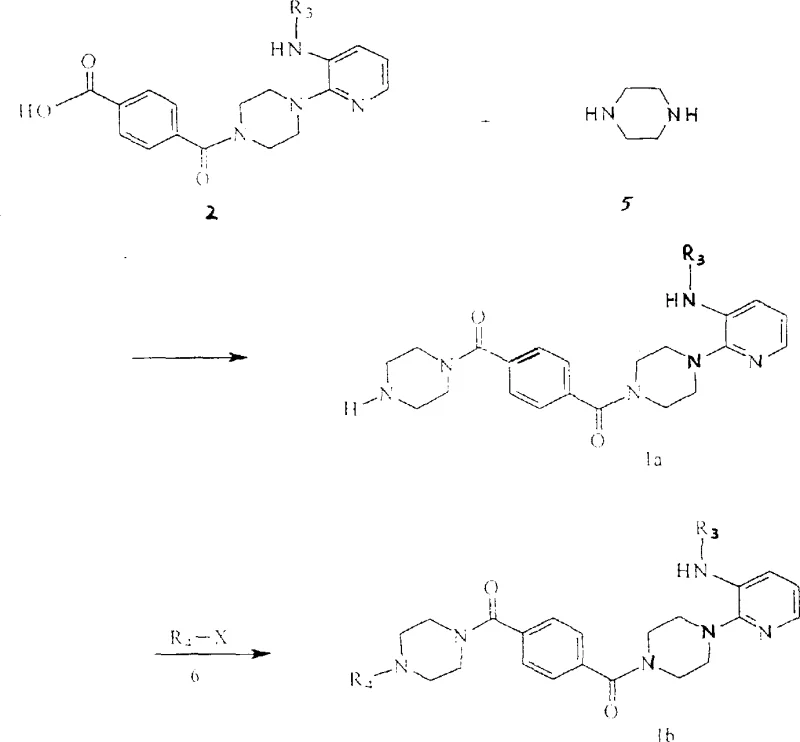
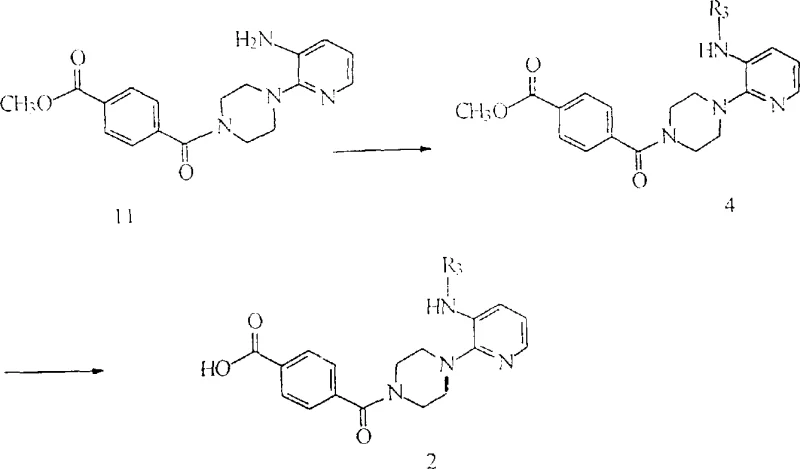
The synthesis of these advanced antiviral intermediates follows a logical and scalable sequence centered around the activation of terephthalic acid derivatives. The process typically begins with the formation of a mixed anhydride using pivaloyl chloride, which serves as a highly reactive acylating agent. This activation step is crucial for driving the subsequent amidation to completion without the need for excessive heat or prolonged reaction times. The resulting activated intermediate is then coupled with various amine components, such as substituted piperazines or amino-alcohols, to construct the final molecular architecture. The versatility of this method allows for the late-stage introduction of diversity, enabling the rapid generation of analog libraries for structure-activity relationship (SAR) studies. Detailed standardized synthetic steps see the guide below.
- Prepare the activated acid intermediate by reacting monomethyl terephthalate with pivalyl chloride in the presence of triethylamine at 0-5°C.
- Couple the activated intermediate with substituted piperazine derivatives under controlled temperatures to form the core terephthalamide scaffold.
- Perform final functionalization via reductive alkylation or hydrolysis depending on the desired R1 and R2 substituents to yield the target compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this terephthalamide-based chemistry offers tangible logistical and financial benefits. The primary advantage lies in the commoditization of raw materials; precursors like terephthalic acid derivatives and simple alkyl amines are produced on a massive global scale, insulating the supply chain from the volatility often seen with specialized nucleoside building blocks. This abundance translates directly into cost reduction in pharmaceutical intermediate manufacturing, as the raw material spend constitutes a smaller fraction of the total COGS. Additionally, the reaction conditions described in the patent, typically ranging from 0°C to ambient temperature, eliminate the need for energy-intensive heating or cryogenic cooling infrastructure, further lowering operational expenditures. The robustness of the chemistry also implies higher throughput and shorter cycle times, enhancing overall plant utilization rates.
- Cost Reduction in Manufacturing: The synthetic route eliminates the need for expensive chiral catalysts and complex protecting group manipulations that are staples of nucleoside synthesis. By utilizing direct amidation and nucleophilic substitution, the process reduces the number of unit operations and solvent consumption. This streamlining leads to substantial cost savings in waste treatment and raw material usage. Furthermore, the high yields reported in the patent examples suggest minimal material loss, maximizing the output per kilogram of input. The avoidance of precious metal catalysts also removes the costly and time-consuming step of heavy metal scavenging, simplifying the downstream processing workflow significantly.
- Enhanced Supply Chain Reliability: The reliance on bulk chemicals ensures that production is not held hostage by the limited capacity of niche suppliers. Since the key intermediates are structurally simple and derived from petrochemical feedstocks, the risk of supply disruption due to agricultural failures or fermentation issues is virtually non-existent. This stability allows for long-term contracting and better inventory planning. Moreover, the synthetic pathway is adaptable; if one specific amine substituent becomes scarce, the modular nature of the synthesis allows for the rapid qualification of alternative suppliers or slight structural modifications without redesigning the entire process, ensuring reducing lead time for high-purity intermediates.
- Scalability and Environmental Compliance: The reactions proceed efficiently in common organic solvents like methylene dichloride, ethanol, and acetonitrile, which are easily recovered and recycled in modern facilities. The absence of toxic heavy metals and the use of stoichiometric reagents rather than catalytic systems simplify the environmental impact assessment. Waste streams are primarily organic and saline, which are easier to treat compared to the complex hazardous waste generated by phosphorylation reactions. This environmental friendliness aligns with increasingly stringent global regulations, reducing the compliance burden and facilitating smoother audits. The process is inherently safe, operating at near-ambient pressures and temperatures, which minimizes the risk of thermal runaways and enhances overall plant safety.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these novel terephthalamide derivatives. These insights are derived directly from the experimental data and claims within the patent documentation, providing a clear picture of the technology's maturity and potential. Understanding these aspects is crucial for stakeholders evaluating the feasibility of integrating this chemistry into their existing pipelines.
Q: What is the primary mechanism of action for these terephthalamide derivatives?
A: These compounds function as non-nucleoside inhibitors that target the reverse transcription step of viral replication, specifically inhibiting both HBV reverse transcriptase and RNaseH activities without the toxicity associated with traditional nucleoside analogs.
Q: Are the starting materials for this synthesis commercially available?
A: Yes, key precursors such as monomethyl terephthalate, pivalyl chloride, and various substituted piperazines are widely available commodity chemicals, ensuring a robust and reliable supply chain for large-scale production.
Q: How does this synthetic route compare to traditional nucleoside synthesis?
A: This route avoids complex sugar chemistry and harsh protection/deprotection steps typical of nucleosides, utilizing milder amide coupling conditions that significantly simplify purification and reduce overall manufacturing costs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Terephthalamide Derivatives Supplier
As the demand for effective non-nucleoside antiviral agents continues to rise, securing a dependable source for high-quality intermediates is critical for drug developers. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific amidation and substitution chemistries required for these terephthalamide derivatives with precision. We adhere to stringent purity specifications and operate rigorous QC labs to ensure that every batch meets the exacting standards required for clinical trial materials and eventual commercial API production. Our commitment to quality ensures that your development timeline remains on track without compromise.
We invite you to collaborate with us to accelerate your antiviral drug development programs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and target specifications. By partnering with NINGBO INNO PHARMCHEM, you gain access to deep process knowledge and a flexible supply chain capable of adapting to your evolving needs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how we can add value to your supply chain.