Advanced Manufacturing of ACC Inhibitor Intermediates: A Breakthrough in Chiral Synthesis and Scalability
Advanced Manufacturing of ACC Inhibitor Intermediates: A Breakthrough in Chiral Synthesis and Scalability
The pharmaceutical industry's relentless pursuit of effective treatments for metabolic disorders has placed Acetyl-CoA carboxylase (ACC) inhibitors at the forefront of therapeutic research. Central to the development of these potent compounds is the efficient and scalable production of key chiral intermediates, specifically (2R)-2-(2-methoxyphenyl)-2-(oxan-4-yloxy)ethane-1-ol. Patent CN111356682A introduces a transformative manufacturing methodology that addresses critical bottlenecks in the synthesis of this vital building block. By shifting away from hazardous reagents and inefficient separation techniques, this novel process offers a robust pathway for the mass production of high-purity intermediates essential for thienopyrimidine and quinazoline derivatives. For R&D directors and procurement specialists, understanding this technological leap is crucial for securing a reliable pharmaceutical intermediate supplier capable of meeting the rigorous demands of modern drug development.
This patent data reveals a sophisticated approach to constructing the chiral center early in the synthesis, thereby avoiding the complexities associated with late-stage resolution. The method leverages a sequence of esterification, hydrolysis, optical resolution via salt formation, and final reduction. This strategic design not only enhances the overall yield but also drastically simplifies the purification workflow. As global demand for ACC inhibitors grows to combat obesity and dyslipidemia, the ability to produce these intermediates with consistent quality and reduced environmental impact becomes a decisive competitive advantage. The following analysis dissects the technical merits and commercial implications of this innovative synthetic route.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those disclosed in WO 2013/071169, present significant hurdles for industrial application. The conventional route typically involves the reaction of 2-methoxybenzaldehyde with S,S-dimethylmethanesulfinyl iodide under sodium hydride (NaH) conditions. This approach is fraught with dangers; sodium hydride is a highly flammable and reactive substance that requires stringent safety protocols, making it less than ideal for large-scale operations. Furthermore, the reliance on unfamiliar and specialized reagents like sulfinyl iodides complicates the supply chain and increases raw material costs. Perhaps most critically, the legacy process suffers from abysmal efficiency, with reported yields as low as 21% during the formation of key intermediates.
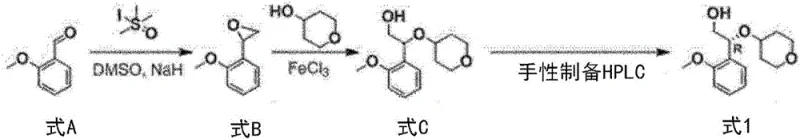
The final purification step in the conventional method relies on Chiral Preparative High-performance liquid chromatography (HPLC) to separate optical isomers. While effective on a laboratory scale, Chiral HPLC is notoriously expensive, time-consuming, and difficult to scale for ton-level production. The mechanical separation of enantiomers using this technology creates a massive bottleneck, limiting throughput and inflating the cost of goods sold (COGS). Additionally, the need for column chromatography to remove impurities further exacerbates waste generation and solvent consumption. For a procurement manager focused on cost reduction in API manufacturing, these inefficiencies represent a substantial financial burden that undermines the commercial viability of the final drug product.
The Novel Approach
In stark contrast, the process outlined in CN111356682A offers a streamlined and economically superior alternative. The new strategy initiates with a substitution reaction between an ethyl ester derivative and tetrahydro-2H-pyran-4-ol, bypassing the need for hazardous hydride reagents entirely. This shift to more benign chemistry significantly lowers the barrier to entry for safe manufacturing. The core innovation lies in the optical resolution step, which utilizes diastereomeric salt formation with (1R,2S)-2-amino-1,2-diphenylethanol. This classical yet highly effective technique allows for the enrichment of the desired (R)-isomer through simple crystallization, completely eliminating the need for costly Chiral HPLC equipment.
By integrating a hydrolysis step followed by a controlled reduction using agents like lithium aluminum hydride or borane dimethyl sulfide, the new process achieves high yields and exceptional purity. The elimination of complex chromatographic separations translates directly into faster cycle times and reduced solvent waste. This approach aligns perfectly with the principles of green chemistry, offering a sustainable solution for the commercial scale-up of complex pharmaceutical intermediates. For supply chain heads, this means a more resilient production line that is less susceptible to the delays and high costs associated with specialized analytical separations, ensuring a steady flow of high-quality materials for downstream synthesis.
Mechanistic Insights into Chiral Resolution and Reduction
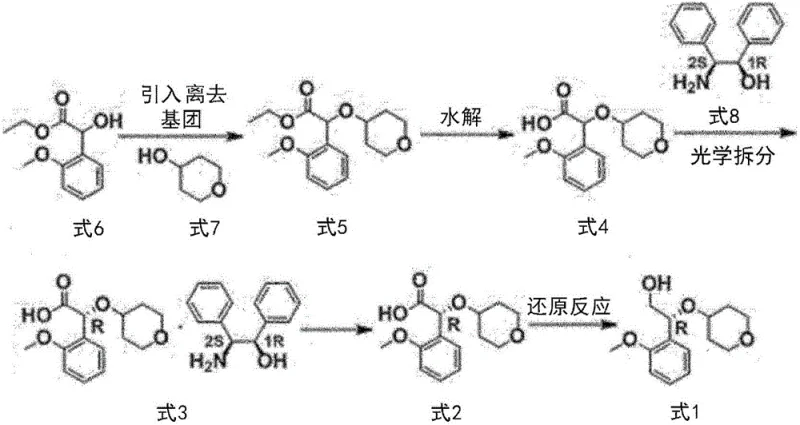
The heart of this novel synthesis lies in the precise control of stereochemistry through diastereomeric salt formation. The process begins with the hydrolysis of the racemic ester (Formula 5) to generate the corresponding carboxylic acid (Formula 4). This acid is then reacted with a chiral amine resolving agent, specifically (1R,2S)-2-amino-1,2-diphenylethanol (Formula 8), in a suitable solvent system such as ethyl acetate. The interaction between the racemic acid and the chiral amine results in the formation of two diastereomeric salts. Due to differences in their physical properties, particularly solubility, one salt preferentially crystallizes out of the solution. This phenomenon allows for the physical separation of the desired enantiomer from its mirror image without the need for chiral columns.
Following the isolation of the optically enriched salt (Formula 3), the free acid (Formula 2) is liberated through acidification and extraction. The final transformation involves the reduction of the carboxylic acid group to a primary alcohol to yield the target compound (Formula 1). The patent specifies the use of potent reducing agents such as lithium aluminum hydride (LiAlH4) or borane dimethyl sulfide (BMS). These reagents are selected for their ability to efficiently reduce the carboxyl group while maintaining the integrity of the sensitive ether linkage and the chiral center. The reaction conditions are carefully optimized, typically operating at temperatures between 0°C to 30°C, to prevent racemization and ensure high optical purity. This mechanistic precision ensures that the final product meets the stringent specifications required for active pharmaceutical ingredients.
Impurity control is inherently built into this workflow. The recrystallization of the diastereomeric salt serves as a powerful purification step, removing not only the unwanted enantiomer but also various organic impurities carried over from previous steps. The subsequent extraction and distillation processes further refine the material. By avoiding the use of transition metal catalysts that often leave trace residues requiring complex removal strategies, this metal-free approach simplifies the impurity profile. For R&D teams, this means a cleaner product with a more predictable stability profile, reducing the risk of failures during regulatory filing and quality control testing.
How to Synthesize (2R)-2-(2-methoxyphenyl)-2-(oxan-4-yloxy)ethane-1-ol Efficiently
The synthesis of this critical ACC inhibitor intermediate is achieved through a logical sequence of four main transformations: substitution, hydrolysis, resolution, and reduction. The process starts with readily available starting materials, ensuring supply chain security. The initial step involves activating the hydroxyl group of the ester precursor with a leaving group, followed by nucleophilic substitution with the pyran alcohol. This is followed by saponification to reveal the acid functionality necessary for resolution. The optical purity is then established through the salt formation described previously, and finally, the carbonyl group is reduced to the alcohol. Detailed standardized synthetic steps see the guide below.
- Perform substitution reaction on ethyl 2-hydroxy-2-(2-methoxyphenyl)acetate with tetrahydro-2H-pyran-4-ol to form the ester intermediate.
- Hydrolyze the ester intermediate using a base such as lithium hydroxide to obtain the racemic acid.
- Conduct optical resolution by forming a salt with (1R,2S)-2-amino-1,2-diphenylethanol and recrystallizing to enhance purity.
- Reduce the resolved chiral acid using lithium aluminum hydride or borane dimethyl sulfide to yield the final alcohol product.
Commercial Advantages for Procurement and Supply Chain Teams
The adoption of the process described in CN111356682A offers profound commercial benefits that extend far beyond the laboratory bench. For procurement managers and supply chain leaders, the shift from the legacy HPLC-based method to this crystallization-driven route represents a significant opportunity for cost reduction in pharmaceutical intermediate manufacturing. The elimination of Chiral Preparative HPLC removes a major capital expenditure and operational cost center. Furthermore, the use of common, commercially available reagents instead of specialized sulfinyl iodides stabilizes raw material pricing and availability. This robustness is essential for maintaining margin integrity in a competitive market.
- Cost Reduction in Manufacturing: The most immediate financial impact comes from the replacement of expensive chromatographic separation with crystallization. Chiral HPLC requires costly columns, high-purity solvents, and significant energy consumption, all of which drive up the unit cost. By switching to salt formation, the process utilizes standard reactors and filtration equipment, leading to substantially lower processing costs. Additionally, the improved yields reported in the patent examples, reaching up to 97% in certain steps compared to the 21% of the prior art, mean that less raw material is wasted per kilogram of finished product. This efficiency gain directly translates to a lower cost of goods sold, allowing for more competitive pricing strategies.
- Enhanced Supply Chain Reliability: Supply chain continuity is often threatened by reliance on niche reagents or complex processing steps. The conventional method's dependence on sodium hydride poses logistical challenges due to its classification as a dangerous good, requiring special shipping and storage. The new process avoids such hazardous materials, simplifying logistics and reducing insurance and compliance costs. Moreover, the resolving agent, (1R,2S)-2-amino-1,2-diphenylethanol, is a well-established chiral pool chemical with a stable global supply. This ensures that production schedules are not disrupted by raw material shortages, providing a reliable pharmaceutical intermediate supplier with the ability to meet long-term contract commitments consistently.
- Scalability and Environmental Compliance: Scaling a process that relies on column chromatography is notoriously difficult and often impractical for multi-ton production. The new method is inherently scalable, utilizing unit operations like stirring, heating, and filtration that are standard in any GMP facility. From an environmental perspective, the reduction in solvent usage associated with eliminating HPLC and column chromatography significantly lowers the facility's E-factor (mass of waste per mass of product). This aligns with increasingly strict environmental regulations and corporate sustainability goals. The ability to run this process in standard stainless steel reactors without specialized linings or containment further facilitates rapid scale-up from pilot plant to commercial production.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this new synthesis route is vital for stakeholders evaluating its implementation. The following questions address common concerns regarding purity, safety, and scalability, drawing directly from the data provided in the patent documentation. These insights are intended to clarify the operational advantages and technical feasibility of the process for potential partners and internal review committees.
Q: How does this new process improve upon the conventional Chiral HPLC method?
A: The new process replaces expensive and low-throughput Chiral Preparative HPLC with a cost-effective diastereomeric salt formation using (1R,2S)-2-amino-1,2-diphenylethanol, significantly reducing production costs and increasing scalability.
Q: What are the safety advantages of this synthesis route?
A: Unlike the prior art which utilizes hazardous sodium hydride (NaH) and unfamiliar sulfinyl iodides, this novel method employs standard reagents like methanesulfonyl chloride and common reducing agents, greatly enhancing operational safety for large-scale manufacturing.
Q: What optical purity can be achieved with this method?
A: Through repeated recrystallization of the diastereomeric salt intermediate, the process achieves high optical purity exceeding 98%, ensuring the quality required for downstream API synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (2R)-2-(2-methoxyphenyl)-2-(oxan-4-yloxy)ethane-1-ol Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful development of ACC inhibitors hinges on the availability of high-quality chiral intermediates. Our technical team has thoroughly analyzed the pathways described in CN111356682A and possesses the expertise to implement this advanced synthesis at scale. We understand that transitioning from lab-scale discovery to commercial manufacturing requires more than just a recipe; it demands rigorous process optimization and quality control. Our facilities are equipped to handle the specific requirements of this chemistry, ensuring that every batch meets the stringent purity specifications demanded by top-tier pharmaceutical companies.
We invite you to leverage our capabilities to accelerate your drug development timeline. With extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, we are positioned to be your strategic partner in supply. Our rigorous QC labs ensure that optical purity and chemical identity are verified at every stage. To discuss how we can support your project with a Customized Cost-Saving Analysis, please contact our technical procurement team today. We are ready to provide specific COA data and route feasibility assessments tailored to your unique volume and quality requirements.