Advanced Thiazolopyrimidine Manufacturing: Enhancing Yield and Purity for Global Pharma Supply Chains
Advanced Thiazolopyrimidine Manufacturing: Enhancing Yield and Purity for Global Pharma Supply Chains
The pharmaceutical industry constantly seeks robust synthetic routes that maximize yield while minimizing impurity profiles, particularly for complex heterocyclic scaffolds like thiazolopyrimidines. Patent CN1914213A introduces a transformative methodology for the preparation of these critical pharmaceutical intermediates, addressing long-standing efficiency bottlenecks in the field. By implementing a strategic nitrogen protection protocol prior to the key amination step, this technology significantly enhances the overall process efficiency compared to conventional direct substitution methods. For R&D directors and procurement specialists, understanding this innovation is vital for securing a reliable pharmaceutical intermediate supplier capable of delivering high-purity materials consistently. The following analysis details how this patented approach optimizes the synthesis of formula (I) compounds, ensuring better resource utilization and supply chain stability.
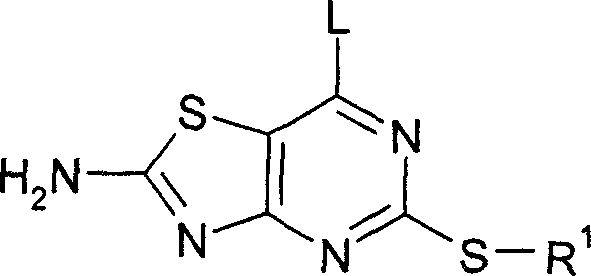
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 7-substituted thiazolo[4,5-d]pyrimidin-2-ones involved the direct nucleophilic displacement of a leaving group, such as chlorine, on the unprotected heterocyclic core. As documented in prior art like WO-01/25242, this direct approach often suffers from significant inefficiencies, primarily due to the reactivity of the thiazole nitrogen. Without protection, the thiazole nitrogen can participate in unwanted side reactions or lead to decomposition pathways during the harsh conditions required for amination. Experimental data from previous methods indicates an overall yield of approximately 40% when converting the chloro-intermediate to the final amino-product. This low efficiency translates to substantial raw material waste and increased purification burdens, creating a bottleneck for cost reduction in API manufacturing. Furthermore, the presence of unprotected nitrogen atoms can complicate the impurity profile, requiring extensive chromatographic purification which is undesirable for large-scale production.
The Novel Approach
The innovative process described in CN1914213A overcomes these limitations by introducing a temporary protecting group on the thiazole nitrogen before the amination step occurs. This strategy stabilizes the heterocyclic core, allowing for cleaner nucleophilic substitution with amines of formula HNR2R3. By shielding the nitrogen, the reaction pathway is directed exclusively towards the desired C-N bond formation at the 7-position, drastically reducing side products. The patent data demonstrates that this modification boosts the overall yield to approximately 70%, representing a near-doubling of efficiency compared to the unprotected route. This improvement is not merely theoretical; it is validated through specific embodiments where intermediates like formula III are isolated and characterized. The ability to isolate stable protected intermediates provides quality control checkpoints that are absent in the one-pot conventional methods, thereby enhancing the reliability of the supply chain for these complex molecules.
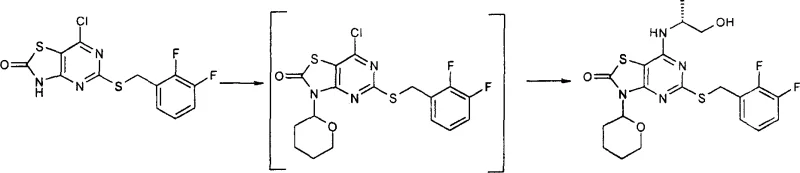
Mechanistic Insights into Nitrogen Protection and Cyclization
The core mechanistic advantage of this process lies in the electronic modulation of the thiazolo[4,5-d]pyrimidine ring system via N-protection. When the thiazole nitrogen is alkylated or silylated with groups such as tetrahydropyranyl (THP) or 2-(phenylsulfonyl)ethyl, the electron density of the ring is altered, rendering the C-7 position more susceptible to nucleophilic attack by the amine while preventing N-alkylation of the thiazole ring itself. This selectivity is crucial for maintaining the structural integrity of the final API intermediate. The protection step typically involves reacting the 7-chloro-thiazolopyrimidinone with a protecting reagent in the presence of an acid catalyst or base, depending on the specific group employed. For instance, the use of 3,4-dihydro-2H-pyran under acidic conditions forms a stable N-THP ether that withstands the subsequent amination temperatures of 60°C to 65°C. This thermal stability ensures that the protecting group remains intact during the displacement reaction, only to be removed in a dedicated final step.
Impurity control is another critical aspect managed by this mechanistic design. In unprotected syntheses, the free NH group can act as a nucleophile or undergo oxidation, leading to dimerization or polymeric byproducts that are difficult to remove. By capping this site, the process effectively eliminates these degradation pathways. The subsequent deprotection step, often achieved through acid hydrolysis or base-assisted elimination (as seen with the phenylsulfonyl ethyl group), is designed to be orthogonal to the rest of the molecule's functionality. For example, the removal of the THP group using dilute hydrochloric acid in acetonitrile allows for the precipitation of the final product directly from the reaction mixture. This crystallization-driven isolation not only simplifies downstream processing but also ensures that the final solid form meets stringent purity specifications required for pharmaceutical applications, minimizing the risk of genotoxic impurities carrying over into the final drug substance.
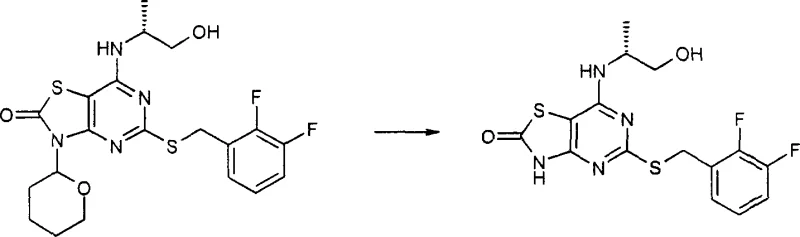
How to Synthesize Thiazolopyrimidine Derivatives Efficiently
The synthesis of these high-value intermediates follows a logical progression from commercially available starting materials, ensuring accessibility and reproducibility. The process begins with the formation of the pyrimidinol core, followed by cyclization to the thiazole-fused system, protection, amination, and finally deprotection. Each step has been optimized to balance reaction kinetics with ease of workup, utilizing common industrial solvents like acetonitrile, toluene, and tetrahydrofuran. The detailed standardized synthesis steps see the guide below, which outlines the specific conditions for maximizing yield and purity at each stage of the transformation. This structured approach allows manufacturers to predictably scale the reaction from laboratory benchtop to pilot plant without encountering unexpected exotherms or solubility issues.
- Prepare the thioether pyrimidinol precursor by reacting 4-amino-6-hydroxy-2-mercaptopyrimidine with a substituted benzyl halide in aqueous THF.
- Cyclize the precursor using chlorocarbonylsulfenyl chloride to form the thiazolo[4,5-d]pyrimidin-2-one core structure.
- Protect the thiazole nitrogen with a suitable group (e.g., THP or phenylsulfonyl ethyl) before performing nucleophilic aromatic substitution with the desired amine.
- Remove the protecting group under acidic or basic conditions to isolate the final high-purity thiazolopyrimidine product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers tangible benefits beyond simple chemical elegance. The primary advantage is the drastic improvement in mass balance; achieving a 70% overall yield compared to the historical 40% baseline means that significantly less raw material is required to produce the same amount of finished goods. This efficiency gain directly correlates to cost reduction in pharmaceutical intermediate manufacturing, as the consumption of expensive reagents like substituted benzyl halides and chiral amines is minimized. Additionally, the robustness of the crystallization steps described in the examples reduces the reliance on resource-intensive chromatography, further lowering the cost of goods sold (COGS). The process is designed to be scalable, utilizing unit operations that are standard in multi-purpose chemical plants, thereby reducing lead time for high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The elimination of low-yielding steps and the avoidance of complex purification techniques result in substantial cost savings. By preventing the formation of hard-to-remove impurities through nitrogen protection, the process reduces the need for multiple recrystallizations or column chromatography, which are major cost drivers in fine chemical production. The use of readily available reagents such as chlorocarbonylsulfenyl chloride and common protecting group reagents ensures that material costs remain stable and predictable. Furthermore, the higher yield reduces the volume of waste solvent and chemical byproducts that require disposal, contributing to lower environmental compliance costs and a smaller carbon footprint for the manufacturing site.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, such as 4-amino-6-hydroxy-2-mercaptopyrimidine and various benzyl halides, are commercially available from multiple global suppliers, mitigating the risk of single-source dependency. The robustness of the reaction conditions, which tolerate moderate variations in temperature and stoichiometry without significant yield loss, ensures consistent batch-to-batch quality. This reliability is critical for maintaining continuous API production schedules and avoiding costly delays. The ability to isolate stable intermediates, such as the N-protected chloro-compounds, allows for the strategic stocking of key precursors, providing a buffer against supply chain disruptions and enabling just-in-time delivery models for downstream customers.
- Scalability and Environmental Compliance: The process is inherently scalable, as demonstrated by the successful execution of reactions on multi-gram scales in the patent examples using standard glassware and heating mantles. The solvent systems employed, primarily acetonitrile and toluene, are easily recoverable and recyclable through distillation, aligning with green chemistry principles. The final isolation steps often involve simple filtration and washing, which are easily adapted to large-scale centrifuges and dryers. This simplicity reduces the complexity of the equipment train required for commercial scale-up of complex pharmaceutical intermediates, lowering capital expenditure for new production lines. Moreover, the reduced generation of hazardous waste due to higher atom economy supports stricter environmental regulations and sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this thiazolopyrimidine synthesis technology. These answers are derived directly from the experimental data and claims within patent CN1914213A, providing a factual basis for decision-making. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this route into their existing manufacturing portfolios. The focus is on practical applicability, yield expectations, and the versatility of the protecting group strategies employed.
Q: How does the nitrogen protection strategy improve yield in thiazolopyrimidine synthesis?
A: By protecting the thiazole nitrogen prior to amination, the process prevents unwanted side reactions and decomposition, increasing overall yield from approximately 40% to 70%.
Q: What protecting groups are suitable for this synthesis route?
A: The patent identifies several effective protecting groups including tetrahydropyranyl (THP), 2-(phenylsulfonyl)ethyl, and methoxymethyl, each removable under specific hydrolytic or basic conditions.
Q: Is this process scalable for commercial API intermediate production?
A: Yes, the method utilizes standard organic solvents like acetonitrile and toluene, and involves robust crystallization steps, making it highly suitable for commercial scale-up from kilograms to metric tons.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Thiazolopyrimidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the competitive landscape of pharmaceutical development. Our team of expert chemists has extensively analyzed the methodology presented in CN1914213A and possesses the technical capability to implement this advanced protection strategy immediately. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale optimization to full-scale manufacturing is seamless and risk-mitigated. Our facilities are equipped with rigorous QC labs and state-of-the-art analytical instrumentation to guarantee that every batch of thiazolopyrimidine intermediate meets stringent purity specifications, fully compliant with international regulatory standards.
We invite potential partners to engage with our technical procurement team to discuss how this improved synthesis route can benefit your specific project requirements. By leveraging our expertise, you can access a Customized Cost-Saving Analysis that quantifies the potential economic impact of switching to this higher-yield process for your supply chain. We encourage you to contact us to request specific COA data for similar heterocyclic intermediates and to receive detailed route feasibility assessments tailored to your target molecules. Let us collaborate to optimize your supply chain and secure a reliable source of high-quality pharmaceutical intermediates for your future success.