Advanced Manufacturing of L-FMAU Intermediates via Novel Electrophilic Fluorination Pathways
Advanced Manufacturing of L-FMAU Intermediates via Novel Electrophilic Fluorination Pathways
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for potent antiviral agents, particularly for the treatment of Hepatitis B Virus (HBV) and Epstein-Barr Virus (EBV). Patent CN1571792A introduces a groundbreaking methodology for the preparation of 2'-deoxy-2'-halo-beta-L-arabinofuranosyl nucleosides, with a specific focus on 1-(2'-deoxy-2'-fluoro-beta-L-arabinofuranosyl)-thymine, commonly known as L-FMAU. This technology represents a significant leap forward in nucleoside chemistry by utilizing L-arabinose, a commercially abundant and cost-effective starting material, rather than the traditionally expensive L-ribose or L-xylose. The process eliminates the need for specialized equipment and relies on inexpensive reagents, making it highly attractive for large-scale commercial production. By streamlining the synthesis into a manageable 10-step reaction sequence, this patent addresses critical supply chain bottlenecks associated with complex carbohydrate modifications.
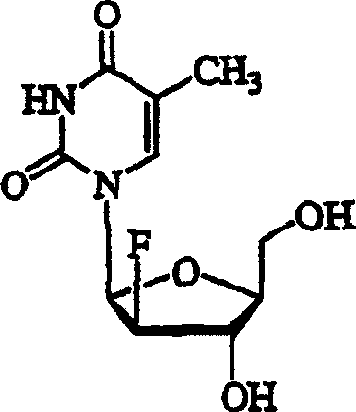
L-FMAU has demonstrated exceptional activity against HBV and EBV, making it a high-value target for API manufacturers. The structural integrity of the 2'-fluoro modification is crucial for metabolic stability and viral polymerase inhibition. The patent details a comprehensive approach that begins with the protection of L-arabinose and proceeds through a series of stereoselective transformations. Unlike previous methods that suffered from low overall yields and harsh reaction conditions, this invention leverages electrophilic fluorination strategies to introduce the critical fluorine atom with high precision. The ability to produce high-purity intermediates without generating excessive toxic byproducts aligns perfectly with modern green chemistry initiatives and regulatory requirements for pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of L-FMAU has been plagued by inefficiencies stemming from the choice of starting materials and the complexity of the reaction pathways. Prior art, such as the methods described by Cheng and Chu, relied heavily on L-xylose or L-ribose as the primary carbon sources. As illustrated in the traditional pathways, converting L-xylose to the necessary intermediates involved a tedious 14-step sequence with a dismal total yield of approximately 8%. Even routes starting from L-ribose, while slightly more direct, still incurred high costs due to the premium price of the starting sugar. Furthermore, these conventional methods often utilized nucleophilic fluorinating agents like DAST (diethylaminosulfur trifluoride), which are notoriously unstable and hazardous to handle on a multi-kilogram scale. The instability of such reagents poses significant safety risks and complicates process validation, leading to increased operational costs and potential supply disruptions.
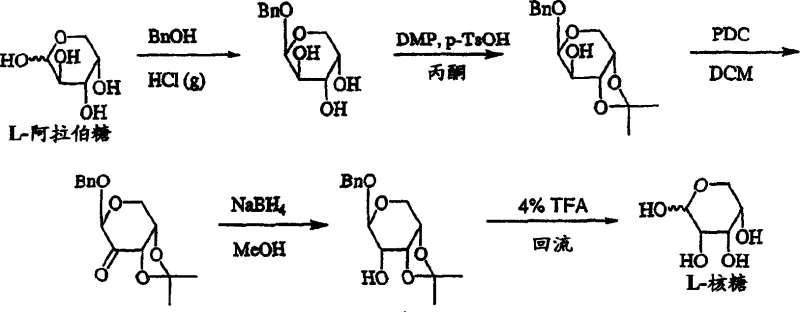
Another major drawback of the legacy processes was the poor stereocontrol during the formation of the furanose ring. The conversion of pyranose precursors to the biologically active furanose form often resulted in complex mixtures of anomers, requiring extensive chromatographic purification that further eroded the overall yield. In some reported cases, the total yield of important intermediates from L-xylose was as low as 20%, rendering the process economically unviable for generic drug manufacturing. The reliance on expensive protecting group strategies, such as multiple benzoylation and acetylation steps without efficient recycling, added to the material costs. Additionally, the use of heavy metal catalysts or difficult-to-remove reagents in older protocols created impurity profiles that were challenging to control, necessitating rigorous and costly downstream processing to meet pharmaceutical purity standards.
The Novel Approach
The methodology disclosed in CN1571792A fundamentally reimagines the synthetic landscape by prioritizing L-arabinose, a pentose sugar that is significantly cheaper and more readily available than its stereoisomers. This novel approach circumvents the need to convert L-arabinose into L-ribose, thereby saving multiple synthetic steps and avoiding the associated yield losses. The core innovation lies in the direct functionalization of the L-arabinose scaffold through a carefully orchestrated sequence of protection, halogenation, and electrophilic fluorination. By employing Selectfluor as the fluorinating agent, the process achieves high regioselectivity for the 2'-position without the safety hazards associated with DAST. This shift from nucleophilic to electrophilic fluorination not only enhances operator safety but also improves the reproducibility of the reaction, a critical factor for technology transfer and commercial scale-up.
Furthermore, the patent introduces a highly efficient method for the critical pyranose-to-furanose isomerization. By utilizing a catalytic amount of sulfuric acid in anhydrous methanol, the process drives the equilibrium towards the desired furanoside with remarkable efficiency, reportedly achieving yields of up to 80% in this specific step. This is a substantial improvement over acid hydrolysis methods that often led to decomposition or incomplete conversion. The streamlined 10-step total synthesis minimizes the number of isolation points, reducing solvent consumption and waste generation. The use of standard laboratory equipment and common solvents like methanol, acetone, and dichloromethane ensures that the process can be implemented in existing manufacturing facilities without the need for capital-intensive retrofitting. This accessibility makes the technology particularly valuable for contract development and manufacturing organizations (CDMOs) looking to offer cost-competitive nucleoside intermediates.
Mechanistic Insights into Electrophilic Fluorination and Isomerization
The heart of this synthetic strategy is the precise introduction of the fluorine atom at the 2'-position of the sugar ring. The mechanism begins with the preparation of L-arabinal derivatives, which serve as the substrate for electrophilic addition. When treated with Selectfluor (F-TEDA-BF4), the electron-rich double bond of the glycal undergoes a concerted addition where the fluorine cation adds to the C2 position. This electrophilic fluorination is superior to nucleophilic displacement because it avoids the formation of elimination byproducts that are common when using fluoride salts on activated leaving groups. The reaction conditions, typically involving a mixture of nitromethane and water or acetone and water, are mild enough to preserve the stereochemical integrity of the adjacent chiral centers while ensuring complete conversion of the starting glycal. The resulting 2-deoxy-2-fluoro-L-arabinopyranose is formed with high diastereoselectivity, setting the stage for the subsequent ring contraction.
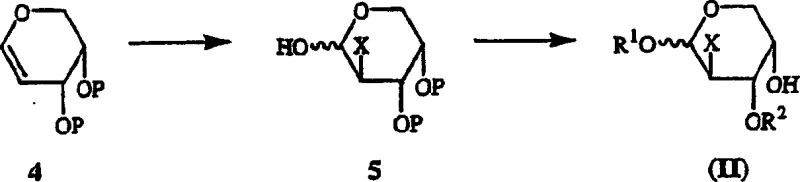
Following fluorination, the conversion of the six-membered pyranose ring to the five-membered furanose ring is a pivotal transformation that determines the final biological activity of the nucleoside. The patent describes a mechanism where the 2-deoxy-2-fluoro-L-arabinopyranose is treated with one equivalent of sulfuric acid in anhydrous methanol under reflux conditions. This acid-catalyzed process facilitates the opening of the pyranose ring to an acyclic oxocarbenium ion intermediate, which then recyclizes to form the thermodynamically favored furanoside. The use of anhydrous methanol serves a dual purpose: it acts as the solvent and provides the methoxy group at the anomeric position, stabilizing the furanose form as a methyl glycoside. This stabilization prevents mutarotation and simplifies the purification of the intermediate. The mechanistic understanding of this equilibrium allows for the optimization of reaction time and temperature, ensuring that the ratio of alpha to beta anomers is controlled effectively before the final coupling with the nucleobase.
Impurity control is rigorously addressed through the selection of protecting groups and reaction conditions. The use of acetyl groups in the early stages provides adequate protection while remaining easy to remove under mild basic conditions, such as sodium methoxide in methanol. Later in the synthesis, benzoyl groups are introduced to enhance the crystallinity of intermediates and facilitate purification via column chromatography or recrystallization. The patent notes that trace amounts of L-ribose isomers may form during fluorination, but these can be managed through careful control of the reaction stoichiometry and workup procedures. The final coupling with thymine, activated via silylation with HMDS (hexamethyldisilazane), proceeds through a standard Vorbrüggen-type mechanism, ensuring the formation of the beta-N-glycosidic bond with high fidelity. This comprehensive mechanistic approach ensures that the final API intermediate meets the stringent purity specifications required for antiviral drug development.
How to Synthesize L-FMAU Efficiently
The synthesis of L-FMAU according to this patent involves a logical progression from cheap bulk chemicals to high-value fine intermediates. The process starts with the peracetylation of L-arabinose, followed by bromination to generate the glycosyl bromide. Reduction of this bromide yields the protected L-arabinal, which is then subjected to the key fluorination step. Subsequent deprotection and isomerization convert the pyranose to the furanose methyl glycoside. Finally, after benzoylation and activation to the bromo-sugar, the molecule is coupled with silylated thymine to yield the protected nucleoside, which is then deprotected to give L-FMAU. This route is designed for scalability, avoiding cryogenic temperatures or ultra-high vacuum conditions that hinder industrial adoption. For detailed standardized operating procedures and specific molar ratios, please refer to the technical guide below.
- Protect L-arabinose and convert to L-arabinal derivative via bromination and zinc reduction.
- Perform electrophilic fluorination using Selectfluor to generate 2-deoxy-2-fluoro-L-arabinopyranose.
- Convert the pyranose to furanose form using sulfuric acid in anhydrous methanol, followed by benzoylation and coupling with thymine.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the shift to L-arabinose as the starting material offers immediate and tangible economic benefits. L-arabinose is a commodity chemical produced in large volumes for the food and fermentation industries, resulting in a stable and low-cost supply base compared to the niche market for L-ribose. This raw material availability significantly reduces the risk of supply chain disruptions caused by vendor shortages or price volatility. By eliminating the need to purchase expensive chiral pool sugars, manufacturers can drastically lower the bill of materials for each batch of L-FMAU produced. Furthermore, the reduction in the total number of synthetic steps from 14 to 10 directly correlates with reduced labor costs, lower utility consumption, and decreased solvent usage. These operational efficiencies translate into a more competitive pricing structure for the final API, allowing pharmaceutical companies to improve their margins or pass savings on to healthcare providers.
Supply chain reliability is further enhanced by the robustness of the chemical reactions described in the patent. The use of stable reagents like Selectfluor and common acids like sulfuric acid means that the process is less sensitive to variations in reagent quality or storage conditions. This robustness simplifies quality control testing for incoming raw materials and reduces the likelihood of batch failures due to reagent degradation. The avoidance of hazardous reagents like DAST also lowers the regulatory burden associated with handling and disposing of toxic chemicals, facilitating faster environmental permitting and site approvals. For global supply chains, this means that the manufacturing process can be replicated across different geographic locations with consistent results, ensuring a continuous supply of critical antiviral intermediates even in the face of regional logistical challenges. The scalability of the process from gram to kilogram scales has been demonstrated in the examples, providing confidence for rapid capacity expansion.
Environmental compliance and waste management are increasingly critical factors in supplier selection. This novel synthesis route generates fewer hazardous byproducts and utilizes solvents that are easier to recover and recycle. The high yields achieved in key steps, such as the 80% yield in the isomerization step and 77% in the final deprotection, mean that less raw material is wasted as unreacted starting material or side products. This atom economy contributes to a lower environmental footprint, aligning with the sustainability goals of major pharmaceutical corporations. The ability to recycle protecting group byproducts, such as converting ribose byproducts back to the desired intermediate, further minimizes waste. For supply chain heads, this translates to reduced costs for waste disposal and a stronger corporate social responsibility profile. The combination of cost reduction, supply security, and environmental stewardship makes this technology a strategic asset for long-term procurement planning.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. Understanding these details is crucial for R&D teams evaluating the feasibility of adopting this route for their own production lines. The answers are derived directly from the experimental data and claims within patent CN1571792A, ensuring accuracy and relevance. These insights help clarify the advantages over legacy methods and provide a clear picture of the operational requirements.
Q: Why is L-arabinose preferred over L-ribose for L-FMAU synthesis?
A: L-arabinose is commercially available at a significantly lower cost compared to L-ribose or L-xylose. The patented method utilizes L-arabinose directly, avoiding the expensive and low-yield conversion steps associated with traditional L-ribose routes.
Q: What is the key advantage of using Selectfluor in this process?
A: Selectfluor enables efficient electrophilic fluorination of the L-arabinal intermediate. This avoids the use of unstable nucleophilic fluorinating agents like DAST and allows for better control over regioselectivity and safety during scale-up.
Q: How does the new method improve overall yield compared to prior art?
A: Conventional methods from L-xylose or L-arabinose via L-ribose reported total yields as low as 8% to 20%. The novel 10-step pathway described in CN1571792A optimizes critical steps like the pyranose-to-furanose conversion, achieving significantly higher efficiency without requiring specialized equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable L-FMAU Supplier
NINGBO INNO PHARMCHEM stands at the forefront of nucleoside intermediate manufacturing, leveraging advanced technologies like the one described in CN1571792A to deliver superior value to our global partners. Our expertise in scaling diverse pathways from 100 kgs to 100 MT/annual commercial production ensures that we can meet the demanding volume requirements of the antiviral market. We possess extensive experience in handling complex carbohydrate chemistry and fluorination reactions, backed by stringent purity specifications and rigorous QC labs that guarantee every batch meets international pharmacopoeia standards. Our commitment to process optimization allows us to offer high-purity L-FMAU intermediates with consistent quality, supporting your drug development timelines and commercial launch goals.
We invite you to collaborate with us to explore how this innovative synthesis route can benefit your specific project needs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating exactly how switching to this L-arabinose-based route can optimize your budget. Please contact our technical procurement team today to request specific COA data for our L-FMAU batches and discuss route feasibility assessments for your next-generation antiviral programs. Together, we can accelerate the delivery of life-saving medications to patients worldwide while maintaining the highest standards of efficiency and quality.