Scalable Synthesis of Lumateperone Intermediates via Novel One-Pot Cyclization Technology
Introduction to Advanced Lumateperone Intermediate Manufacturing
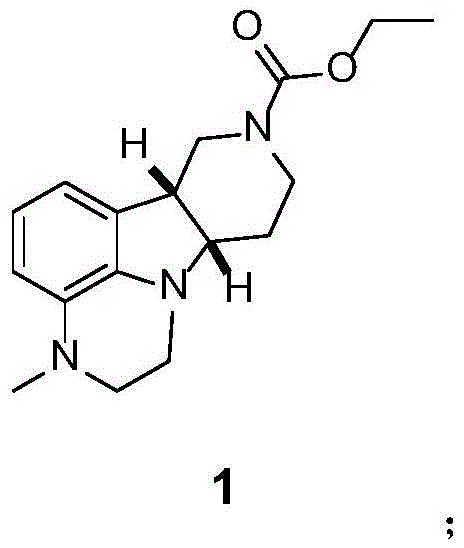
The pharmaceutical landscape for schizophrenia treatment has been significantly altered by the approval of Lumateperone, necessitating robust and scalable supply chains for its complex intermediates. Patent CN113024554A discloses a groundbreaking preparation method for the key intermediate (6bR,10aS)-2,3,6b,9,10,10a-hexahydro-3-methyl-1H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxaline-8(7H)-carboxylic acid ethyl ester, addressing critical bottlenecks in prior art. This technical disclosure represents a paradigm shift from labor-intensive, low-yield processes to a streamlined, high-efficiency protocol that leverages early-stage chiral resolution and a novel one-pot cyclization strategy. For global procurement and R&D teams, understanding this methodology is essential for securing a reliable pharmaceutical intermediates supplier capable of meeting the stringent quality and volume demands of modern antipsychotic drug production.
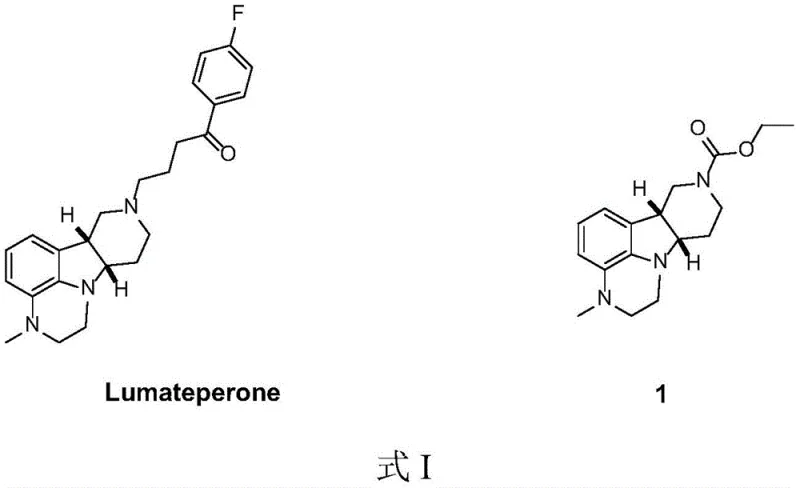
The structural complexity of Lumateperone requires precise stereochemical control, which has historically been a major hurdle in commercial manufacturing. The patent highlights that existing methods often suffer from poor atom economy, reliance on precious metal catalysts, and difficult purification steps that drive up the cost of goods sold. By optimizing the synthesis of the tricyclic core shown in the formula, this new approach not only improves overall yield but also enhances the safety profile of the manufacturing process. As we delve into the technical specifics, it becomes clear that this innovation offers a viable pathway for cost reduction in pharmaceutical intermediates manufacturing, making it a highly attractive candidate for technology transfer and commercial scale-up.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art synthesis routes for Lumateperone intermediates are plagued by inefficiencies that render them suboptimal for large-scale industrial application. For instance, the route described in WO 2000077022 relies on a late-stage chiral separation via preparative liquid chromatography, which typically results in a dismal recovery yield of only 30-40%, effectively wasting more than half of the synthesized material. Furthermore, other methods such as those found in WO 2008112280 utilize borane reductions that present severe safety hazards due to exothermic reactions and gas release during quenching, complicating reactor design and operational safety protocols. Additionally, routes employing palladium-catalyzed couplings, as seen in literature from J Med Chem, introduce expensive catalysts and ligands that increase raw material costs and require rigorous heavy metal removal steps to meet regulatory limits. These cumulative factors create a fragile supply chain vulnerable to cost volatility and production delays.
The Novel Approach
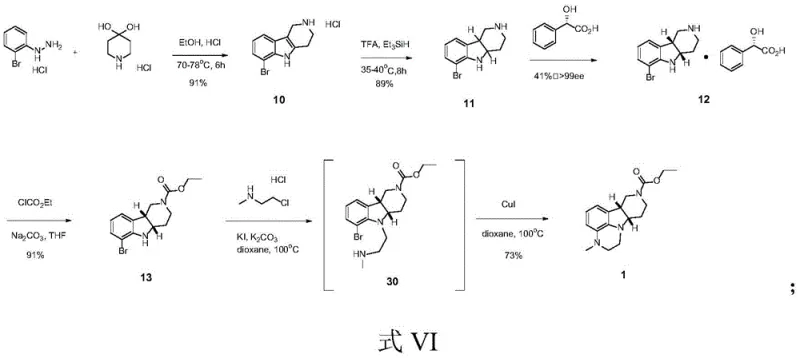
The methodology presented in CN113024554A overcomes these historical deficiencies through a cleverly designed sequence that prioritizes early chirality introduction and convergent ring formation. Instead of deferring stereochemical resolution to the final product, this process resolves the intermediate at the hexahydro-indole stage using (S)-(+)-mandelic acid, achieving optical purity greater than 99% before the final cyclization. The cornerstone of this innovation is the one-pot transformation of the bromo-carbamate precursor into the final tricyclic ester using 2-chloro-N-methylethylamine hydrochloride and a copper(I) iodide catalyst. This single operation accomplishes both N-alkylation and intramolecular cyclization simultaneously, eliminating the need for isolation of unstable intermediates and reducing solvent consumption. The result is a robust process with a reported yield of 73% for the key step and an overall profile that is significantly more amenable to commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into CuI-Catalyzed Cyclization
The core chemical innovation lies in the copper-catalyzed cascade reaction that constructs the piperazine-fused pyrrolo-quinoxaline skeleton. Mechanistically, this involves the initial nucleophilic substitution of the bromine atom on the indole core by the amine nitrogen of 2-chloro-N-methylethylamine, facilitated by the base potassium carbonate. Subsequently, the cuprous iodide catalyst activates the secondary amine formed in situ or facilitates the intramolecular N-arylation to close the third ring, forming the rigid tricyclic structure essential for biological activity. This dual-functionality step is critical because it avoids the formation of linear byproducts that are common in stepwise alkylations. The use of dioxane or DMF as solvents provides the necessary polarity to solubilize the inorganic salts while maintaining the stability of the organometallic catalytic species. Understanding this mechanism allows process chemists to fine-tune reaction parameters such as temperature and catalyst loading to maximize throughput and minimize impurity formation.
Impurity control is another vital aspect addressed by this mechanistic design. In traditional routes, the use of protecting groups like benzophenone imine often leads to difficult-to-remove urea or amide byproducts upon deprotection. By utilizing an ethyl carbamate protecting group that remains stable throughout the cyclization and is part of the final ester functionality, the process inherently reduces the generation of foreign organic impurities. Furthermore, the early resolution step ensures that diastereomeric impurities are removed before the most complex bond-forming reactions occur, preventing the amplification of stereochemical errors. This strategic placement of purification checkpoints ensures that the final high-purity pharmaceutical intermediate meets the rigorous specifications required for subsequent coupling with the fluorobenzophenone side chain, ultimately safeguarding the quality of the final API.
How to Synthesize Lumateperone Intermediate Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for reproducing this high-yielding process in a GMP environment. The sequence begins with a classic Fischer-type indole synthesis to establish the tetracyclic framework, followed by a selective reduction and resolution to set the stereochemistry. The final stages involve protection and the pivotal copper-catalyzed cyclization. Each step has been optimized for yield and purity, with specific attention paid to workup procedures that avoid column chromatography, favoring crystallization and extraction instead. For detailed operational parameters including exact stoichiometry, temperature ramps, and quenching protocols, please refer to the standardized guide below which encapsulates the critical process parameters defined in the intellectual property.
- Condense 2-bromophenylhydrazine hydrochloride with 4-piperidone hydrochloride monohydrate in ethanol under acidic reflux to form the tetracyclic core (Compound 10).
- Reduce the double bond of Compound 10 using trifluoroacetic acid and triethylsilane to generate the hexahydro-indole derivative (Compound 11).
- Perform chiral resolution of Compound 11 using (S)-(+)-mandelic acid in ethanol to isolate the optically pure (4aS,9bR) enantiomer (Compound 12).
- Protect the secondary amine of Compound 12 with ethyl chloroformate to yield the carbamate precursor (Compound 13).
- Execute a one-pot reaction of Compound 13 with 2-chloro-N-methylethylamine hydrochloride using potassium carbonate and cuprous iodide to finalize the tricyclic structure (Compound 1).
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthesis route offers transformative benefits for supply chain stability and cost management. The elimination of expensive palladium catalysts and the avoidance of late-stage chiral HPLC separation directly translate to a lower cost of goods sold, allowing for more competitive pricing in the generic and branded drug markets. Moreover, the replacement of hazardous borane reductions with a silane-based system mitigates safety risks, reducing insurance premiums and facility upgrade costs associated with handling pyrophoric reagents. These factors combined create a manufacturing process that is not only economically superior but also operationally resilient against regulatory and safety audits.
- Cost Reduction in Manufacturing: The process achieves significant cost savings by replacing precious metal catalysts with abundant copper salts and eliminating atom-uneconomical protecting groups. By consolidating the alkylation and cyclization into a single one-pot operation, the method drastically reduces solvent usage, energy consumption for heating and cooling cycles, and labor hours required for intermediate isolations. This lean manufacturing approach ensures that the production of the intermediate remains economically viable even at fluctuating raw material prices, providing a sustainable margin structure for long-term supply agreements.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials such as 2-bromophenylhydrazine and 4-piperidone ensures that the supply chain is not dependent on niche or single-source vendors. The robustness of the crystallization-based purification steps, as opposed to chromatography, means that production batches are less likely to fail quality control due to column overload or resolution issues. This predictability allows procurement managers to forecast lead times with greater accuracy, reducing lead time for high-purity pharmaceutical intermediates and ensuring continuous availability for downstream API synthesis.
- Scalability and Environmental Compliance: The simplified workflow with fewer unit operations inherently generates less waste, aligning with green chemistry principles and reducing the burden on wastewater treatment facilities. The absence of heavy metal contaminants from palladium catalysts simplifies the environmental compliance dossier, accelerating regulatory approvals for new drug master files. Furthermore, the mild reaction conditions and the use of standard organic solvents facilitate easy technology transfer from pilot plant to multi-ton commercial reactors, ensuring that the pharmaceutical intermediates supplier can rapidly ramp up capacity to meet market demand without compromising on quality or safety standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. These answers are derived directly from the comparative data and experimental examples provided in the patent documentation, offering clarity on how this method outperforms legacy technologies in terms of yield, purity, and operational safety.
Q: Why is the new one-pot cyclization method superior to previous palladium-catalyzed routes?
A: Previous methods, such as those described in J Med Chem (2014), relied on expensive palladium catalysts and ligands along with atom-uneconomical protecting groups like benzophenone imine. The new method utilizes a cost-effective copper(I) iodide catalyst and eliminates the need for bulky protecting groups, significantly reducing raw material costs and simplifying downstream purification.
Q: How does this process address the challenges of chiral purity in Lumateperone synthesis?
A: Traditional routes often deferred chiral separation to the final step, requiring expensive preparative chiral HPLC with low recovery yields (30-40%). This patented approach introduces an early-stage resolution using (S)-(+)-mandelic acid after the reduction step, achieving optical purity greater than 99% before the final cyclization, thereby ensuring high stereochemical integrity throughout the synthesis.
Q: What are the safety advantages of this route regarding reduction steps?
A: Earlier synthetic pathways utilized borane-tetrahydrofuran complexes for reduction, which pose significant safety risks due to exothermic heat release and gas evolution during quenching, making large-scale operations hazardous. The disclosed method employs a triethylsilane/trifluoroacetic acid system, which offers a milder, more controllable reduction profile suitable for industrial amplification without the severe thermal hazards associated with borane reagents.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Lumateperone Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient intermediate synthesis in the value chain of antipsychotic medications. Our technical team has thoroughly analyzed the pathway described in CN113024554A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this innovative process to life. We are committed to delivering products with stringent purity specifications and have invested in rigorous QC labs equipped with advanced chiral HPLC and NMR capabilities to ensure every batch meets the highest global standards. Our facility is designed to handle complex heterocyclic chemistry safely, ensuring that the transition from lab scale to commercial supply is seamless and compliant with all international regulatory requirements.
We invite potential partners to engage with our technical procurement team to discuss how this optimized route can benefit your specific supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this copper-catalyzed method. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project timelines, ensuring that your development programs proceed without interruption due to material shortages.