Advanced Synthesis of Chiral 3-(Aminomethyl)-3-fluoro-2-indolone Derivatives for Pharmaceutical Applications
The pharmaceutical industry continuously seeks efficient routes to fluorinated heterocycles due to their profound impact on metabolic stability and bioavailability. A significant breakthrough in this domain is documented in patent CN107056676B, which discloses a robust method for preparing chiral 3-(aminomethyl)-3-fluoro-2-indolone derivatives. These structures serve as critical scaffolds for bioactive molecules, notably including Maxi-K potassium channel openers like BMS-204352. Traditional synthetic pathways often suffer from cumbersome activation steps and limited substrate tolerance, creating bottlenecks in drug discovery pipelines. This innovative technology overcomes these hurdles by employing a direct nucleophilic addition strategy using chiral (R)-N-(tert-butylsulfinyl)imines, commonly known as Ellman imines. By leveraging the stereochemical control inherent in the sulfinyl group, the method achieves exceptional diastereoselectivity under mild conditions. For global procurement teams and R&D directors, this represents a pivotal shift towards more reliable pharmaceutical intermediate supplier capabilities, ensuring consistent access to high-value chiral building blocks essential for next-generation therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this invention, the synthesis of 3-(aminomethyl)-3-fluoro-2-indolones was severely constrained by the necessity of using highly activated and difficult-to-prepare precursors. Existing literature primarily described addition reactions involving 3-fluoro-3-(2,2,2-trifluoro-1,1-dihydroxyethyl)-2-indolone, a substrate that requires complex multi-step preparation and handling. Furthermore, the scope of compatible imines in these legacy methods was restricted almost exclusively to polyfluoroalkylaldimines, drastically limiting the chemical diversity accessible to medicinal chemists. This lack of versatility meant that introducing varied aryl, heteroaryl, or alkyl groups at the 3-position was often impractical or impossible without redesigning the entire synthetic route. Additionally, the harsh conditions often associated with activating the indolone core could lead to decomposition or racemization, compromising the optical purity required for potent biological activity. These inefficiencies translate directly into higher costs and longer lead times for cost reduction in API manufacturing, as extensive purification and specialized reagents become necessary to overcome the inherent limitations of the starting materials.
The Novel Approach
The methodology outlined in the patent introduces a streamlined and universally applicable solution by utilizing unactivated 3-fluoro-2-indolone directly as the nucleophile. In this elegant transformation, the 3-fluoro-2-indolone is deprotonated in situ by a strong base to generate a reactive enolate species, which then attacks the electrophilic carbon of the chiral sulfinylimine. This approach eliminates the need for pre-functionalized indolone derivatives, significantly simplifying the supply chain and reducing raw material costs. The reaction proceeds efficiently in common organic solvents such as tetrahydrofuran (THF), diethyl ether, or dichloromethane at temperatures ranging from -80°C to 0°C. As demonstrated in the typical reaction scheme below, the process tolerates a wide array of substituents on the imine component, including phenyl, substituted phenyl, naphthyl, and heteroaryl groups.
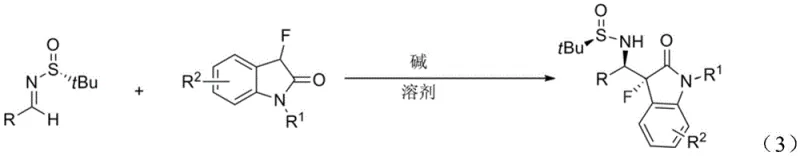
The versatility of this method allows for the rapid generation of diverse libraries of chiral indolinones, facilitating faster structure-activity relationship (SAR) studies. Moreover, the use of commercially available tert-butylsulfinamides as the chiral source ensures that the process is not dependent on proprietary or exotic catalysts, enhancing its viability for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Asymmetric Nucleophilic Addition
The success of this synthesis relies heavily on the precise stereochemical control exerted by the chiral auxiliary. The reaction initiates with the deprotonation of the acidic proton at the C3 position of the 3-fluoro-2-indolone by a strong non-nucleophilic base such as LiHMDS or KHMDS. This generates a planar or rapidly equilibrating enolate intermediate that is poised for nucleophilic attack. The chirality is induced by the bulky tert-butylsulfinyl group attached to the nitrogen of the imine electrophile. This group creates a rigid chiral environment that shields one face of the imine double bond, forcing the incoming nucleophile to attack from the less hindered trajectory. The structural integrity of the chiral imine, as depicted in the general formula below, is crucial for maintaining high diastereomeric ratios (dr).

In the transition state, coordination between the lithium cation (from the base) and the oxygen atoms of both the sulfinyl group and the indolone carbonyl likely organizes the reactants into a highly ordered cyclic arrangement. This chelation control minimizes background non-selective reactions and ensures that the newly formed C-C bond is established with high fidelity. Following the addition, the resulting adduct retains the sulfinyl group, which can subsequently be removed under acidic conditions to yield the free amine if desired, or retained as a protected functionality depending on the downstream synthetic requirements. This mechanistic robustness explains the consistently high dr values observed across various substrates, often exceeding 99:1, which is critical for meeting the stringent purity specifications demanded by regulatory bodies for high-purity pharmaceutical intermediates.
How to Synthesize Chiral 3-(Aminomethyl)-3-fluoro-2-indolone Efficiently
Executing this synthesis requires careful attention to moisture exclusion and temperature control to maximize yield and stereoselectivity. The protocol involves dissolving the 3-fluoro-2-indolone and the chiral imine in anhydrous solvent under an inert nitrogen atmosphere. A solution of the base is then added dropwise at low temperature to prevent exothermic runaway and minimize side reactions. After the reaction period, typically lasting between 0.5 to 6 hours, the mixture is quenched carefully with aqueous ammonium chloride. The product is isolated via standard liquid-liquid extraction and purified by flash column chromatography. For detailed operational parameters and specific stoichiometric ratios optimized for different substrates, please refer to the standardized guide below.
- Prepare the reaction mixture by combining chiral (R)-N-(tert-butylsulfinyl)imine and 3-fluoro-2-indolone in an anhydrous organic solvent such as THF or diethyl ether under inert atmosphere.
- Cool the reaction mixture to a temperature between -80°C and 0°C, then slowly add a strong base such as LiHMDS, NaHMDS, or tBuOK dropwise to initiate deprotonation.
- Stir the reaction at low temperature for 0.5 to 6 hours, quench with aqueous ammonium chloride, extract with ethyl acetate, and purify via flash column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, this technology offers compelling advantages that address common pain points in the supply of complex chiral intermediates. By shifting away from activated, unstable precursors to stable, commodity-grade starting materials, the process inherently reduces supply chain risk and volatility. The elimination of specialized activation steps translates directly into fewer unit operations, lower energy consumption, and reduced waste generation, all of which contribute to a more sustainable and cost-effective manufacturing profile. Furthermore, the broad substrate scope means that a single platform technology can be used to produce a wide variety of analogues, allowing suppliers to respond rapidly to changing project needs without requalifying entirely new processes.
- Cost Reduction in Manufacturing: The use of economically accessible starting materials, such as simple aldehydes and commercial tert-butylsulfinamide, drastically lowers the raw material cost basis compared to methods requiring custom-synthesized activated indolones. The reaction conditions are mild and do not require expensive transition metal catalysts or ligands, removing the need for costly metal scavenging steps and heavy metal testing. This simplification of the bill of materials and processing requirements leads to substantial overall cost savings, making the production of these high-value intermediates more financially viable for large-scale applications.
- Enhanced Supply Chain Reliability: Because the key reagents are commercially available and the reaction does not rely on air-sensitive or pyrophoric catalysts that degrade upon storage, the supply chain is significantly more robust. The process tolerates a range of solvents including THF, ether, and DCM, providing flexibility in sourcing and logistics. The high yields and excellent diastereoselectivity reported in the patent examples minimize the need for recycling or reprocessing off-spec material, ensuring a consistent and reliable flow of product to meet tight project timelines and reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The reaction operates at manageable low temperatures (-80°C to 0°C) which are achievable with standard industrial cooling systems, facilitating easy scale-up from laboratory to pilot and commercial plant scales. The workup procedure involves a simple aqueous quench and extraction, avoiding complex distillations or hazardous reagents that complicate waste treatment. This aligns well with modern green chemistry principles and environmental regulations, reducing the burden of waste disposal and ensuring long-term operational compliance for manufacturing facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis route. Understanding these details is vital for process chemists evaluating the feasibility of adopting this method for their specific pipeline candidates. The answers are derived directly from the experimental data and technical disclosures within the patent documentation.
Q: What are the key advantages of this synthesis method over prior art?
A: Unlike previous methods requiring activated 3-fluoro-3-(2,2,2-trifluoro-1,1-dihydroxyethyl)-2-indolone, this novel approach utilizes readily available 3-fluoro-2-indolone and chiral Ellman imines directly. It offers broader substrate scope, milder reaction conditions, and superior diastereoselectivity (dr up to 99:1).
Q: Which bases are suitable for this asymmetric addition reaction?
A: The patent specifies several strong non-nucleophilic bases that effectively promote the reaction, including lithium bis(trimethylsilyl)amide (LiHMDS), sodium bis(trimethylsilyl)amide (NaHMDS), potassium bis(trimethylsilyl)amide (KHMDS), sodium tert-butoxide, and potassium tert-butoxide.
Q: Is this process scalable for commercial API intermediate production?
A: Yes, the process is highly scalable. It employs economical starting materials, operates at manageable low temperatures (-80°C to 0°C), and utilizes standard workup procedures (aqueous quench and extraction), making it suitable for kilogram-to-ton scale manufacturing of high-purity pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral 3-(Aminomethyl)-3-fluoro-2-indolone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality chiral building blocks play in accelerating drug development. Our team of expert process chemists has extensively evaluated the technology described in CN107056676B and possesses the capability to implement this route effectively. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from gram-scale discovery to multi-ton manufacturing. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including chiral HPLC analysis to guarantee the optical integrity of every batch we deliver.
We invite you to collaborate with us to leverage this advanced synthesis technology for your next program. By partnering with our technical procurement team, you can obtain a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in chiral fluorinated intermediates can drive efficiency and value for your organization.