Advanced Solid-Phase Synthesis Strategy for High-Purity Enfuvirtide Production
The pharmaceutical landscape for HIV treatment continues to evolve, demanding more efficient and cost-effective manufacturing routes for critical antiretroviral agents. Patent CN102241746B introduces a robust methodology for the preparation of Enfuvirtide, also known as T-20, a pivotal fusion inhibitor used in the management of chronic human immunodeficiency virus infections. This intellectual property outlines a comprehensive solid-phase peptide synthesis (SPPS) strategy that fundamentally shifts the production paradigm from traditional fragment condensation to a streamlined, full-sequence assembly. By leveraging specific resin supports and optimizing the acidolysis cleavage cocktail, the disclosed technique addresses long-standing challenges regarding purity, yield, and operational complexity. For stakeholders in the pharmaceutical intermediates sector, this patent represents a significant technological leap, offering a pathway to produce high-purity polypeptides with enhanced economic viability and reduced environmental footprint compared to legacy processes.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of complex polypeptides like Enfuvirtide relied heavily on a hybrid approach combining liquid-phase and solid-phase fragment condensation. This conventional methodology, while functional, suffered from inherent inefficiencies that hampered large-scale commercialization. The process necessitated the independent synthesis and subsequent purification of multiple peptide fragments, each requiring rigorous isolation steps that dramatically extended the production cycle. Furthermore, the reliance on specific acidolysis solvents containing dithiothreitol (DTT) introduced a severe economic bottleneck; DTT is an exceptionally expensive reagent, with market values historically exceeding 20,000 RMB per kilogram, rendering the process cost-prohibitive for mass production. Additionally, the use of diverse solvent systems and the inability to achieve continuous automatic production resulted in inconsistent batch quality, typically capping purity levels between 93% and 95%, which is suboptimal for modern regulatory standards requiring ultra-high purity profiles for injectable biologics.
The Novel Approach
In stark contrast, the novel approach detailed in the patent utilizes a complete sequence solid-phase synthesis technique anchored on specialized aminoresins such as Rink Amide AM resin. This methodology eliminates the need for fragment condensation entirely, allowing for the linear assembly of the entire 36-amino acid sequence directly on the solid support. A critical innovation lies in the substitution of the cleavage cocktail components; the process replaces the costly DTT with 1,2-ethanedithiol (EDT), a commercially abundant scavenger available at a fraction of the cost—approximately one-twentieth the price of DTT—while delivering superior or equivalent scavenging performance. This strategic reagent swap, combined with a unified solvent system primarily based on DMF, facilitates fully automatic production capabilities. The result is a simplified workflow that not only enhances operational safety by avoiding toxic hydrogen fluoride but also drives the final product purity to exceed 98%, establishing a new benchmark for cost reduction in peptide manufacturing.
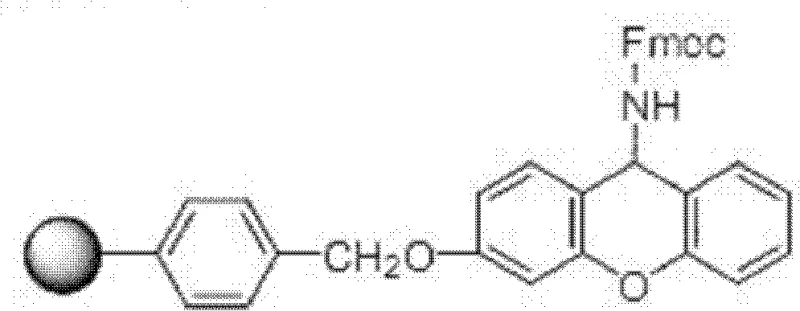
Mechanistic Insights into Fmoc-Based Solid-Phase Peptide Synthesis
The core of this synthesis relies on the Fmoc (9-fluorenylmethyloxycarbonyl) protection strategy, which offers distinct advantages over Boc chemistry, particularly regarding the mildness of the deprotection conditions. The process initiates with the coupling of Fmoc-Phe to the aminoresin, forming a stable amide linkage that serves as the anchor for the growing peptide chain. As illustrated in the structural representations of the resin-bound intermediates, the choice of linker is paramount; the patent highlights the efficacy of Rink Amide-type linkers which allow for the generation of C-terminal amides upon cleavage, matching the native structure of Enfuvirtide. The coupling reactions are mediated by potent condensing agents such as N,N'-diisopropylcarbodiimide (DIC) in the presence of activating additives like 1-hydroxybenzotriazole (HOBt). This combination ensures rapid activation of the carboxyl group of the incoming Fmoc-amino acid, minimizing racemization and maximizing coupling efficiency to near-quantitative levels (>99%) at each step, which is critical for preventing the accumulation of deletion sequences in such a long peptide.
Following the initial loading, the synthesis proceeds through iterative cycles of deprotection and coupling. The removal of the Fmoc group is achieved using a piperidine/DMF solution, a standard yet highly effective protocol that exposes the free amine for the next coupling event without disturbing the side-chain protecting groups (such as tBu, Boc, or Trt). The structural diversity of the resins available, including variations like the Sieber resin shown below, provides flexibility in tuning the acid sensitivity of the linker. This mechanistic precision allows for the controlled elongation of the peptide chain from the C-terminus to the N-terminus. The final acetylation of the N-terminus is performed on-resin, ensuring that the terminal modification is complete before the harsh cleavage conditions are applied. This on-resin capping strategy prevents the formation of unacetylated impurities, further simplifying the downstream purification burden and enhancing the overall homogeneity of the crude peptide prior to HPLC processing.
How to Synthesize Enfuvirtide Efficiently
The execution of this synthesis requires precise control over stoichiometry and reaction times to ensure high fidelity across all 36 coupling cycles. The protocol dictates a molar excess of Fmoc-amino acids (2.5 to 3.5 equivalents relative to resin loading) to drive the equilibrium towards product formation, coupled with reaction times optimized between 100 to 140 minutes for difficult couplings. The detailed standardized synthetic steps, including specific washing protocols and reagent concentrations, are essential for reproducibility. For a comprehensive breakdown of the exact operational parameters and safety precautions required for laboratory or pilot-scale execution, please refer to the technical guide below.
- Couple Fmoc-Phe to aminoresin (e.g., Rink Amide AM) using DIC and HOBt to form the initial resin-bound intermediate.
- Perform sequential coupling of Fmoc-protected amino acids from position 2 to 36, removing Fmoc groups with piperidine/DMF between steps.
- Acetylate the N-terminus, then cleave the peptide from the resin using a TFA/EDT/water mixture, followed by HPLC purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this patented synthesis route offers tangible strategic benefits that extend beyond mere technical feasibility. The primary driver of value is the radical optimization of the bill of materials (BOM). By eliminating the dependency on high-cost reagents like DTT and replacing them with commodity chemicals like EDT, the direct material cost per kilogram of active pharmaceutical ingredient is significantly lowered. Furthermore, the shift to a fully automated solid-phase process reduces labor intensity and minimizes the risk of human error, leading to more consistent batch-to-batch quality. This reliability is crucial for maintaining a steady supply of high-purity HIV fusion inhibitors in a market where demand stability is paramount. The simplified workflow also translates to shorter lead times, as the elimination of intermediate fragment purifications accelerates the overall production timeline from raw materials to finished goods.
- Cost Reduction in Manufacturing: The substitution of expensive scavengers and the adoption of a single-solvent system (DMF) for the majority of the synthesis drastically reduce raw material expenditure. The avoidance of complex fragment isolation steps further cuts down on solvent consumption and waste disposal costs, creating a leaner manufacturing profile that improves margin potential without sacrificing quality standards.
- Enhanced Supply Chain Reliability: The use of widely available Fmoc-protected amino acids and standard resins mitigates the risk of supply bottlenecks associated with specialized custom fragments. The robustness of the SPPS method ensures that production can be scaled up or down rapidly in response to market fluctuations, providing a resilient supply base for global pharmaceutical partners seeking a reliable enfuvirtide intermediate supplier.
- Scalability and Environmental Compliance: The process is inherently scalable, having been designed with automatic production in mind, which facilitates the transition from gram-scale R&D to multi-kilogram commercial batches. Additionally, the avoidance of toxic hydrogen fluoride and the use of safer acidolysis cocktails align with modern green chemistry principles, reducing the regulatory burden associated with hazardous waste management and improving the overall sustainability profile of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, aiming to clarify the operational advantages and quality outcomes of the described method. Understanding these nuances is vital for technical teams evaluating the feasibility of adopting this route for their own production pipelines.
Q: How does this patent improve the cost efficiency of Enfuvirtide production?
A: The method replaces expensive dithiothreitol (DTT) with 1,2-ethanedithiol (EDT) in the acidolysis step. Since EDT costs significantly less than DTT while maintaining effective scavenging properties, the overall raw material cost is drastically reduced without compromising product quality.
Q: What purity levels can be achieved with this solid-phase synthesis route?
A: By utilizing a full-sequence solid-phase synthesis strategy combined with optimized HPLC purification conditions (using sodium acetate/acetonitrile systems), the process consistently yields Enfuvirtide with purity exceeding 98%, addressing the limitations of older fragment condensation methods.
Q: Is this process suitable for large-scale automated manufacturing?
A: Yes, the protocol is designed for automation. It uses a single solvent system (DMF) for most coupling steps and avoids complex fragment purifications, making it highly compatible with automated peptide synthesizers for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Enfuvirtide Supplier
The technological advancements detailed in patent CN102241746B underscore the potential for highly efficient, cost-effective production of complex polypeptides like Enfuvirtide. At NINGBO INNO PHARMCHEM, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent supply of materials that meet stringent purity specifications. Our rigorous QC labs are equipped to validate every batch against the high standards set forth in this patent, guaranteeing that the final API or intermediate performs reliably in downstream formulation processes. We are committed to bridging the gap between innovative patent chemistry and commercial reality.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific supply chain requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this method. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments tailored to your volume needs, ensuring a seamless integration of our high-quality intermediates into your manufacturing workflow.