Advanced Synthesis of Alpha-Alpha-Fluorochloroarylmethyl Phosphonates for Enzyme Inhibitor Development
Introduction to Novel Alpha-Alpha-Fluorochloroarylmethyl Phosphonates
The landscape of medicinal chemistry is constantly evolving with the discovery of new scaffolds that offer enhanced biological activity and metabolic stability. Patent CN101492472A introduces a significant breakthrough in this domain by disclosing a novel class of alpha,alpha-fluorochloroarylmethyl phosphonates, represented by the general formula (I). These compounds are not merely structural analogs but represent a sophisticated evolution of alpha-fluorophosphonates, which have long been recognized for their ability to mimic phosphate groups in biological systems. The unique incorporation of both fluorine and chlorine atoms at the alpha-position adjacent to the phosphonate moiety creates a distinct electronic environment that enhances their potential as Protein Tyrosine Phosphatases (PTPs) inhibitors. This structural motif is particularly valuable for researchers developing therapeutics for complex diseases such as cancer, diabetes, and obesity, where PTPs play a critical regulatory role. The patent provides a robust and scalable methodology for accessing these high-value intermediates, addressing a previous gap in the literature where such specific alpha,alpha-disubstituted derivatives were unreported.
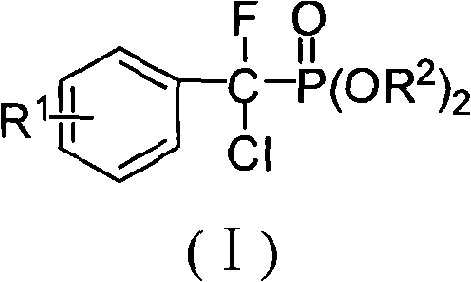
For procurement specialists and supply chain managers in the fine chemical sector, the emergence of this technology signals a new opportunity for sourcing reliable pharmaceutical intermediates supplier materials. The synthetic route described is designed with scalability in mind, utilizing commercially available starting materials such as substituted benzaldehydes and diethyl phosphite. This accessibility is crucial for maintaining cost reduction in pharmaceutical intermediates manufacturing, as it avoids the need for exotic or prohibitively expensive reagents. Furthermore, the method's reliance on standard organic transformations ensures that the production process can be easily adapted to existing manufacturing infrastructure, thereby reducing lead time for high-purity pharmaceutical intermediates. As we delve deeper into the technical specifics, it becomes evident that this patent offers a comprehensive solution for the commercial scale-up of complex enzyme inhibitor precursors, balancing high yield with operational simplicity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of alpha-heteroatom substituted phosphonates has been fraught with challenges related to regioselectivity and reaction control. Conventional methods often rely on direct halogenation or fluorination of pre-formed phosphonates, which can lead to a mixture of mono- and di-substituted products, complicating downstream purification. Specifically, introducing a fluorine atom at the alpha-position without affecting other sensitive functional groups on the aromatic ring requires extremely specific conditions that are difficult to maintain on a large scale. Traditional approaches might utilize hazardous reagents like elemental fluorine or aggressive Lewis acids, posing significant safety risks and environmental compliance issues for modern chemical plants. Moreover, the lack of a chlorine atom in many conventional alpha-fluorophosphonates limits their utility as versatile building blocks, as the chlorine serves as a valuable leaving group for further derivatization into amines or other pharmacophores. These limitations often result in lower overall yields and higher production costs, making the final active pharmaceutical ingredients less economically viable for widespread therapeutic use.
The Novel Approach
In stark contrast, the methodology outlined in CN101492472A presents a streamlined, three-step sequence that elegantly overcomes these historical hurdles. By first establishing the carbon-phosphorus bond through a Pudovik addition, the process ensures that the phosphonate group is securely attached before any halogenation occurs. This sequential logic allows for the independent optimization of each substitution event. The subsequent conversion of the alpha-hydroxyl group to a chlorine atom using triphenylphosphine and carbon tetrachloride is a classic yet highly effective transformation that proceeds with excellent fidelity. Finally, the introduction of the fluorine atom via electrophilic substitution using N-fluorobisbenzenesulfonamide (NFSI) represents a state-of-the-art application of modern fluorination chemistry. This approach not only guarantees the specific alpha,alpha-disubstitution pattern required for PTPs inhibition but also tolerates a wide range of substituents on the aromatic ring, including nitro, cyano, and methoxy groups. This versatility is a key driver for cost reduction in specialty chemical manufacturing, as a single platform technology can generate a diverse library of analogs for structure-activity relationship (SAR) studies.
Mechanistic Insights into the Three-Step Synthetic Sequence
The core of this innovation lies in the precise orchestration of three distinct chemical mechanisms, each contributing to the final structural integrity of the molecule. The journey begins with the Pudovik addition reaction, where an aromatic aldehyde reacts with a dialkyl phosphite in the presence of a base such as sodium ethoxide. This nucleophilic addition forms a tetrahedral intermediate that collapses to yield the alpha-hydroxy arylmethyl phosphonate (II). The reaction is typically conducted in dichloromethane at low temperatures, around -35°C, to suppress side reactions and ensure high stereochemical purity. This step is fundamental as it sets the stage for the subsequent substitutions, creating a chiral center that can be manipulated in later stages. The efficiency of this addition is critical, as demonstrated by yields reaching up to 96% in optimized examples, providing a robust foundation for the rest of the synthesis.
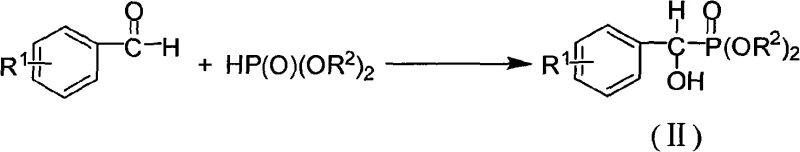
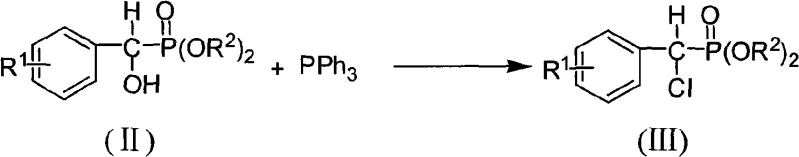
Following the formation of the hydroxyl intermediate, the process moves to a nucleophilic substitution reaction to install the chlorine atom. This transformation utilizes triphenylphosphine (PPh3) in carbon tetrachloride (CCl4) under reflux conditions. Mechanistically, this resembles an Appel reaction, where the hydroxyl group is activated by the phosphine-halogen complex and subsequently displaced by a chloride ion. This step is crucial for converting the relatively poor leaving group (hydroxyl) into a good leaving group (chloride), while simultaneously introducing the first halogen atom. The use of CCl4 as both solvent and chlorine source simplifies the reaction setup, although modern green chemistry adaptations might seek alternative solvents. The resulting alpha-chloro arylmethyl phosphonate (III) is isolated in high yields, often exceeding 80%, demonstrating the reliability of this chlorination protocol for various substituted benzaldehydes.
The final and perhaps most sophisticated step is the electrophilic fluorination to generate the target alpha,alpha-fluorochloro structure. This reaction employs N-fluorobisbenzenesulfonamide (NFSI) as the fluorine source and sodium hexamethyldisilazide (NaHMDS) as a strong, non-nucleophilic base. The reaction is performed in anhydrous tetrahydrofuran (THF) at cryogenic temperatures, initially at -78°C and gradually warming to -30°C. The base deprotonates the acidic alpha-proton adjacent to the phosphonate and chlorine, generating a carbanion that is stabilized by the electron-withdrawing groups. This nucleophilic carbon then attacks the electrophilic fluorine atom of the NFSI reagent. The strict temperature control is vital to prevent decomposition of the reactive intermediates and to ensure selective mono-fluorination. This mechanistic precision allows for the successful synthesis of diverse derivatives, including those with electron-withdrawing nitro and cyano groups, which might otherwise be sensitive to harsher fluorination conditions.
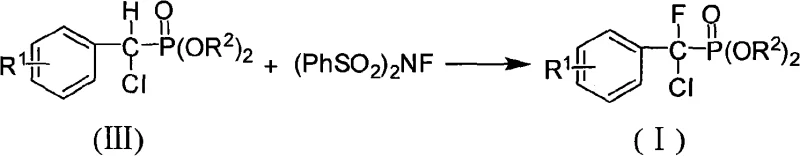
How to Synthesize Alpha-Alpha-Fluorochloroarylmethyl Phosphonate Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific operational parameters to maximize yield and safety. The process begins with the preparation of the alpha-hydroxy intermediate, where moisture control is paramount to prevent hydrolysis of the phosphite reagent. Operators must ensure that the addition of the aldehyde to the phosphite-base mixture is done slowly to manage the exotherm effectively. Once the hydroxyl compound is secured, the chlorination step demands careful handling of carbon tetrachloride and triphenylphosphine, with adequate ventilation due to the volatility of the solvent. The final fluorination step is the most technically demanding, requiring a reactor capable of maintaining temperatures as low as -78°C and the safe handling of strong bases like NaHMDS. For a detailed breakdown of the standardized operating procedures and safety protocols, please refer to the guide below.
- Perform Pudovik addition of aromatic aldehyde and phosphite in dichloromethane with sodium ethoxide at -35°C to form alpha-hydroxy arylmethyl phosphonate.
- Conduct nucleophilic substitution using triphenylphosphine in carbon tetrachloride under reflux to convert the hydroxyl group to a chlorine atom.
- Execute electrophilic fluorination using N-fluorobisbenzenesulfonamide (NFSI) and NaHMDS base in THF at low temperatures (-78°C to -30°C).
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers substantial strategic advantages for organizations looking to secure a reliable supply of high-value enzyme inhibitor intermediates. The primary benefit lies in the significant cost optimization achieved through the use of commodity chemicals. Starting materials such as substituted benzaldehydes and diethyl phosphite are produced on a multi-ton scale globally, ensuring price stability and consistent availability. This contrasts sharply with routes that depend on custom-synthesized or rare organometallic reagents, which are subject to volatile pricing and supply disruptions. By leveraging this technology, procurement managers can negotiate better contracts and reduce the overall cost of goods sold (COGS) for their downstream drug development projects. Furthermore, the high yields reported in the patent examples translate directly to reduced waste generation and lower disposal costs, aligning with modern sustainability goals.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts, which are often required in cross-coupling reactions for similar structures, results in a drastic simplification of the purification process. Without the need for expensive scavengers to remove trace metals, the downstream processing becomes more efficient and less costly. Additionally, the high atom economy of the Pudovik addition step ensures that the majority of the starting mass is incorporated into the final product, minimizing raw material waste. This efficiency is compounded by the ability to recycle solvents like dichloromethane and THF, further driving down operational expenditures. Consequently, the overall manufacturing footprint is leaner, allowing for competitive pricing in the global market for pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The reliance on robust, well-understood chemical transformations mitigates the risk of batch failures that can plague more exotic synthetic methods. Each step in the sequence has been validated across a wide range of substrates, from electron-rich methoxy derivatives to electron-deficient nitro compounds, proving the method's generality. This robustness ensures that suppliers can maintain consistent production schedules, reducing the lead time for high-purity pharmaceutical intermediates. For supply chain heads, this predictability is invaluable, as it allows for accurate inventory planning and reduces the need for safety stock. The ability to source these intermediates from multiple qualified manufacturers who can adopt this standard protocol further strengthens supply security.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from gram-scale laboratory synthesis to kilogram and ton-scale commercial production. The reaction conditions, while requiring temperature control, do not necessitate extreme pressures or specialized equipment beyond standard glass-lined or stainless steel reactors. From an environmental standpoint, the avoidance of heavy metals and the use of stoichiometric reagents that generate manageable byproducts (such as triphenylphosphine oxide) simplify waste treatment. This ease of compliance with environmental regulations facilitates faster regulatory approval for manufacturing sites, accelerating the time-to-market for new drug candidates. The process design supports the commercial scale-up of complex pharmaceutical intermediates without compromising on safety or ecological standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these novel phosphonates. Understanding these details is essential for R&D directors evaluating the feasibility of incorporating these intermediates into their drug discovery pipelines. The answers provided are derived directly from the experimental data and technical specifications outlined in the patent documentation, ensuring accuracy and relevance. Whether you are concerned about impurity profiles, reaction scalability, or specific substrate compatibility, this section aims to provide clarity. We encourage technical teams to review these points closely to assess the alignment of this technology with their specific project requirements.
Q: What is the primary application of alpha,alpha-fluorochloroarylmethyl phosphonates?
A: These compounds serve as potent Protein Tyrosine Phosphatases (PTPs) inhibitors and phosphate mimetics, showing potential in treating cancer, diabetes, and neurological diseases.
Q: Why is the three-step synthesis route preferred over direct fluorination?
A: The stepwise approach via alpha-hydroxy and alpha-chloro intermediates allows for precise control over stereochemistry and substitution patterns, avoiding the harsh conditions and low selectivity of direct methods.
Q: What are the critical reaction conditions for the final fluorination step?
A: The final step requires strict temperature control between -78°C and -30°C using NaHMDS as a strong base and NFSI as the fluorinating agent in anhydrous THF to ensure high yield and purity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Alpha-Fluorochloroarylmethyl Phosphonate Supplier
As the demand for sophisticated enzyme inhibitors continues to rise, partnering with an experienced CDMO is critical for translating patent innovations into commercial reality. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our facilities are equipped with state-of-the-art rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of alpha-alpha-fluorochloroarylmethyl phosphonate meets the highest international standards. We understand the complexities involved in handling fluorinated intermediates and have optimized our processes to deliver high-quality materials that accelerate your drug development timelines. Our commitment to quality and reliability makes us the preferred partner for leading pharmaceutical companies worldwide.
We invite you to engage with our technical procurement team to discuss how we can support your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain insights into how our manufacturing efficiencies can translate into tangible budget benefits for your organization. We are prepared to provide specific COA data and route feasibility assessments tailored to your target molecules. Let us collaborate to bring your next-generation PTPs inhibitors from the bench to the clinic, leveraging our expertise in fine chemical synthesis to drive your success.