Advanced Synthesis of Anticancer Semicarbazide Derivatives for Commercial Scale-Up
The pharmaceutical landscape is undergoing a paradigm shift towards targeted therapies, driven by the urgent need to overcome the limitations of traditional cytotoxic agents. In this context, patent CN110156672B presents a groundbreaking advancement in the synthesis of semicarbazide compounds, a class of molecules demonstrating potent in vitro antitumor activity against critical cancer cell lines including H460, Hela, and MCF-7. This technical disclosure outlines a robust, three-step synthetic pathway that not only achieves high yields but also prioritizes operational simplicity and cost-effectiveness, addressing the core challenges faced by modern pharmaceutical intermediate suppliers. By leveraging a novel condensation strategy followed by hydrazinolysis and urea formation, this method provides a scalable solution for producing high-value oncology scaffolds. For R&D directors and procurement specialists alike, understanding the mechanistic nuances and commercial implications of this patent is essential for securing a competitive edge in the development of next-generation anticancer drugs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex heterocyclic intermediates for oncology applications has been plagued by inefficient multi-step sequences that suffer from poor atom economy and hazardous reaction conditions. Traditional routes often rely on heavy metal catalysts or extreme temperatures that complicate purification and introduce toxic residues, posing significant risks to both product quality and environmental compliance. Furthermore, conventional methods for constructing substituted pyridine rings frequently result in low regioselectivity, generating difficult-to-separate isomers that drastically reduce the overall yield of the target molecule. These inefficiencies translate directly into inflated production costs and extended lead times, creating bottlenecks for cost reduction in API manufacturing. The reliance on scarce or expensive starting materials in older methodologies further exacerbates supply chain vulnerabilities, making it difficult for manufacturers to guarantee consistent availability of critical intermediates for clinical and commercial programs.
The Novel Approach
In stark contrast, the methodology described in CN110156672B introduces a streamlined approach that circumvents these historical hurdles through intelligent molecular design and optimized reaction engineering. The process initiates with a highly efficient cyclization reaction using abundant feedstocks like ethyl acetoacetate and ammonium acetate, eliminating the need for exotic reagents. By carefully controlling the molar ratios—specifically employing a slight excess of ethyl acetoacetate (1:1.1)—the inventors have successfully driven the equilibrium towards the desired product, achieving yields between 70% and 85% in the first step alone. This strategic optimization continues through the subsequent hydrazinolysis and isocyanate addition steps, where the use of common solvents like ethanol and glacial acetic acid ensures that the process remains environmentally benign and economically attractive. This novel approach effectively democratizes access to high-purity semicarbazide derivatives, enabling a more agile response to market demands for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Pyridine Ring Construction and Urea Formation
The core of this synthetic innovation lies in the initial construction of the bi-pyridine scaffold, which serves as the structural backbone for the bioactive semicarbazide. The first step involves a condensation reaction where 1-(3-pyridyl)-3-(dimethylamino)-2-propen-1-one reacts with ethyl acetoacetate in the presence of ammonium acetate. Mechanistically, ammonium acetate serves as a convenient source of ammonia, facilitating the cyclization to form the new pyridine ring fused to the existing pyridine substituent. This reaction is conducted under reflux in glacial acetic acid for 5 hours, a condition that provides sufficient thermal energy to overcome the activation barrier while maintaining a homogeneous reaction medium. The subsequent workup involves pouring the reaction mixture into ice water and extracting with ethyl acetate, a standard liquid-liquid extraction technique that effectively separates the organic product from inorganic salts and polar byproducts. The crude product is then purified via silica gel column chromatography using a petroleum ether and ethyl acetate mixture (3:1 v/v), a critical step that ensures the removal of trace impurities before proceeding to the next stage.
Following the formation of the ester intermediate, the pathway proceeds through a nucleophilic acyl substitution known as hydrazinolysis. Here, the ester group is attacked by hydrazine hydrate, displacing the ethoxy group to form the corresponding hydrazide. The patent specifies a significant molar excess of hydrazine hydrate (1:8.1 ratio), which is a deliberate kinetic strategy to ensure complete conversion of the ester, pushing the yield to an impressive 80%-85%. The final transformation involves the reaction of this hydrazide with a substituted isocyanate, specifically 2,4-difluorophenyl isocyanate in the preferred embodiment. This step forms the semicarbazide linkage (-NH-CO-NH-) through a nucleophilic addition of the hydrazide nitrogen to the electrophilic carbon of the isocyanate group. Refluxing in ethanol for 3 hours facilitates this addition, resulting in the final target molecule with yields reaching 80-90%. The structural integrity and substitution pattern of the final product are crucial for its observed biological activity, as depicted in the general structure below.
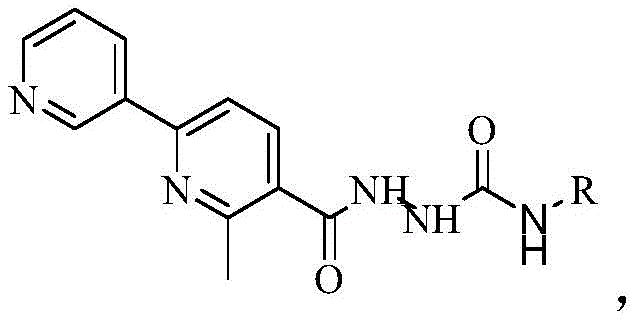
The introduction of the 2,4-difluorophenyl group at the R position is not arbitrary; fluorine substitution is a well-established medicinal chemistry tactic to enhance metabolic stability and membrane permeability. The specific arrangement of the fluorine atoms influences the electronic distribution of the aromatic ring, potentially optimizing the binding affinity of the semicarbazide moiety to its biological target. This level of structural precision underscores the importance of maintaining strict control over reaction stoichiometry and purification protocols throughout the synthesis. For R&D teams, replicating these mechanistic conditions is vital to achieving the reported IC50 values, which indicate potent inhibition of cancer cell proliferation. The ability to systematically vary the 'R' group while maintaining the robustness of the core synthesis offers a versatile platform for structure-activity relationship (SAR) studies, accelerating the discovery of more potent analogues.
How to Synthesize 4-(2,4-difluorophenyl)-1-(2-methyl-6-(pyridin-3-yl)nicotinyl)semicarbazide Efficiently
Executing this synthesis requires strict adherence to the optimized parameters outlined in the patent to ensure reproducibility and high purity. The process is divided into three distinct operational stages, each requiring specific attention to temperature control, reagent addition rates, and workup procedures. The initial cyclization sets the foundation for the entire sequence, making the purification of the intermediate ester critical for downstream success. Operators must ensure that the reflux conditions are maintained consistently to drive the reaction to completion without degrading the sensitive pyridine rings. Following the isolation of the hydrazide, the final coupling with the isocyanate must be monitored closely, typically via TLC, to prevent the formation of urea byproducts from moisture contamination. Detailed standard operating procedures (SOPs) for each step are essential for technology transfer from the laboratory to the pilot plant.
- Condense 1-(3-pyridyl)-3-(dimethylamino)-2-propen-1-one with ethyl acetoacetate and ammonium acetate in glacial acetic acid under reflux to form the pyridine ester intermediate.
- Perform hydrazinolysis of the ester intermediate using hydrazine hydrate in ethanol under reflux to generate the corresponding hydrazide.
- React the hydrazide with substituted isocyanate (e.g., 2,4-difluorophenyl isocyanate) in ethanol under reflux to obtain the final semicarbazide target product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition from laboratory curiosity to commercial viability is often the most significant hurdle. This patented process addresses several critical pain points associated with the sourcing of oncology intermediates. By utilizing commodity chemicals such as ethyl acetoacetate, ammonium acetate, and ethanol, the method significantly reduces the dependency on specialized, high-cost reagents that are prone to supply disruptions. This reliance on bulk chemicals translates directly into a more stable and predictable cost structure, allowing for better long-term budget planning. Furthermore, the simplified purification strategy, which avoids complex distillation or chromatography in the later steps, reduces the consumption of solvents and stationary phases, contributing to substantial cost savings in raw material expenditure.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of ambient pressure reflux conditions drastically lower the capital and operational expenditures associated with production. Traditional methods often require inert atmospheres or cryogenic cooling, which demand specialized equipment and higher energy consumption. In contrast, this process operates under standard heating conditions, making it compatible with existing glass-lined or stainless-steel reactors found in most multipurpose chemical plants. The high yields reported in each step (cumulatively leading to a robust overall yield) mean that less raw material is wasted, directly improving the cost-of-goods-sold (COGS) profile for the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis are widely available from multiple global suppliers, mitigating the risk of single-source dependency. Ethyl acetoacetate and hydrazine hydrate are produced at massive scales for various industries, ensuring a continuous supply even during market fluctuations. This abundance allows procurement teams to negotiate better pricing and secure longer-term contracts, stabilizing the supply chain for high-purity pharmaceutical intermediates. Additionally, the robustness of the reaction conditions means that minor variations in raw material quality can be tolerated without catastrophic failure of the batch, adding a layer of resilience to the manufacturing process.
- Scalability and Environmental Compliance: As regulatory pressures regarding waste disposal intensify, the environmental profile of a synthetic route becomes a key decision factor. This method generates primarily aqueous and organic solvent waste that can be treated using standard effluent treatment protocols, avoiding the generation of heavy metal sludge or hazardous halogenated byproducts common in other halogenation or coupling reactions. The simplicity of the workup—often involving simple filtration and washing—reduces the volume of wastewater generated per kilogram of product. This 'green chemistry' aspect not only lowers disposal costs but also aligns with the sustainability goals of major pharmaceutical companies, facilitating faster regulatory approval and market acceptance.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear picture of what partners can expect when adopting this route. Understanding these details is crucial for assessing the feasibility of integrating this intermediate into your existing drug development pipeline.
Q: What are the key advantages of this semicarbazide synthesis route compared to traditional methods?
A: The patented method (CN110156672B) offers significantly higher yields (up to 90% in the final step) and utilizes readily available raw materials like ethyl acetoacetate and ammonium acetate. Unlike traditional cytotoxic drug synthesis which often involves harsh conditions and complex purification, this route uses mild reflux conditions in common solvents like ethanol and acetic acid, drastically simplifying the operational complexity and reducing waste generation.
Q: How does the impurity profile of this process impact downstream API manufacturing?
A: The process incorporates a silica gel column chromatography purification step after the initial cyclization, ensuring the intermediate ethyl 2-methyl-6-(pyridyl-3-yl)acetate is obtained with high purity. This rigorous control at the intermediate stage prevents the carryover of side products into the final semicarbazide, resulting in a cleaner crude product that requires less intensive recrystallization, thereby enhancing the overall efficiency of the supply chain for high-purity pharmaceutical intermediates.
Q: Is this synthesis method scalable for industrial production of oncology intermediates?
A: Yes, the methodology is explicitly designed for industrial applicability. The reactions rely on standard unit operations such as reflux, filtration, and solvent extraction, avoiding the need for specialized high-pressure equipment or cryogenic conditions. The use of excess reagents like hydrazine hydrate (molar ratio 1:8.1) drives the reaction to completion efficiently, making the commercial scale-up of complex heterocyclic intermediates feasible and economically viable.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Semicarbazide Compound Supplier
The synthesis of 4-(2,4-difluorophenyl)-1-(2-methyl-6-(pyridin-3-yl)nicotinyl)semicarbazide represents a significant opportunity for the development of novel anticancer therapeutics, yet translating this patent into a commercial reality requires a partner with deep technical expertise and manufacturing capacity. NINGBO INNO PHARMCHEM stands ready to support your projects with our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific reflux and filtration requirements of this process, ensuring that every batch meets stringent purity specifications. With our rigorous QC labs and commitment to quality assurance, we guarantee that the intermediates supplied are free from critical impurities, safeguarding the integrity of your downstream API synthesis.
We invite you to collaborate with us to leverage this innovative chemistry for your oncology portfolio. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this route can optimize your overall production economics. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you accelerate your timeline to market with a reliable, scalable, and cost-effective supply of this critical semicarbazide intermediate.