Scalable Synthesis of Furo-Piperidine Derivatives for Kinase Inhibitor Manufacturing
Scalable Synthesis of Furo-Piperidine Derivatives for Kinase Inhibitor Manufacturing
The pharmaceutical industry is constantly seeking robust and cost-effective pathways to access complex heterocyclic scaffolds, particularly those serving as core structures for kinase inhibitors. Patent CN103374005A introduces a transformative methodology for the synthesis of substituted furo-piperidine derivatives, addressing critical bottlenecks in traditional manufacturing. This novel approach leverages furfural, a biomass-derived platform chemical that is both inexpensive and abundantly available, as the primary starting material. By orchestrating a sequence of condensation, reduction, and Pictet-Spengler reactions, the process delivers high-purity intermediates suitable for the production of potent JAK kinase inhibitors. The strategic shift from scarce raw materials to commodity chemicals represents a significant leap forward in process chemistry, offering a viable solution for the commercial scale-up of complex pharmaceutical intermediates.
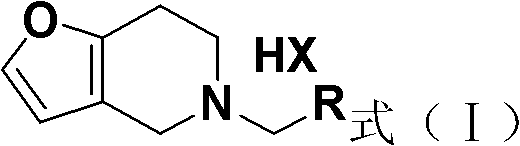
For R&D directors and process chemists, the structural versatility of the final compound, represented as Formula (I), is paramount. The ability to easily modify the R group, typically a phenyl or substituted phenyl moiety, allows for rapid SAR (Structure-Activity Relationship) exploration without overhauling the entire synthetic route. This flexibility is crucial during the lead optimization phase of drug discovery, where speed and adaptability are key drivers of success. Furthermore, the final product can be isolated directly from the reaction system as a salt, such as the hydrochloride, which simplifies the isolation protocol and enhances the overall yield profile.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of substituted furan and piperidine derivatives has been plagued by economic and operational inefficiencies. Conventional procedural styles often rely on 3-furfural as the starting raw material, which is not only significantly more expensive than standard furfural but also suffers from limited supply chain stability. Beyond the raw material costs, the legacy synthesis routes typically necessitate multistep column chromatography purifications to achieve acceptable purity levels. In a commercial setting, column purification is a major bottleneck; it is labor-intensive, solvent-heavy, and difficult to scale beyond kilogram quantities. These factors collectively drive up the cost of goods sold (COGS) and extend the lead time for high-purity pharmaceutical intermediates, making the conventional approach unsustainable for large-volume manufacturing.
The Novel Approach
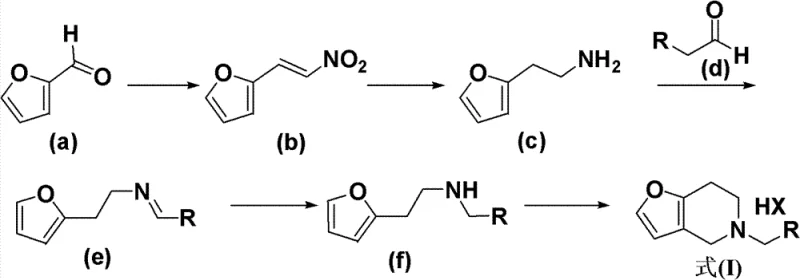
The methodology disclosed in the patent circumvents these challenges through a streamlined, linear synthesis that prioritizes operational simplicity and cost efficiency. As illustrated in the comprehensive reaction scheme below, the process initiates with the condensation of furfural and nitromethane, followed by a reduction to the corresponding amine. This amine then undergoes condensation with an aldehyde and subsequent reduction to form a key secondary amine intermediate. The culmination of the sequence is an elegant Pictet-Spengler cyclization that constructs the fused ring system in a single pot. This route eliminates the need for expensive starting materials and replaces complex chromatographic separations with straightforward crystallization or extraction techniques.
By adopting this novel approach, manufacturers can achieve a drastic reduction in processing time and solvent consumption. The use of common industrial solvents such as methanol, ethanol, and toluene ensures that the process is compatible with existing infrastructure in most fine chemical plants. Moreover, the high atom economy of the condensation and cyclization steps minimizes waste generation, aligning the synthesis with modern green chemistry principles. This makes the technology not only economically superior but also environmentally compliant, a dual advantage that is increasingly valued by global supply chain stakeholders.
Mechanistic Insights into Pictet-Spengler Cyclization
The cornerstone of this synthetic strategy is the final ring-closing step, known as the Pictet-Spengler reaction. This transformation involves the acid-catalyzed condensation of a beta-arylethylamine (in this case, the furan-containing amine) with an aldehyde (formaldehyde) to form an iminium ion intermediate. Under acidic conditions, the electron-rich furan ring acts as a nucleophile, attacking the electrophilic iminium carbon to close the six-membered piperidine ring. The choice of acid, such as HCl, HBr, or p-toluenesulfonic acid, is critical for driving the equilibrium towards the cyclized product while preventing polymerization or degradation of the sensitive furan moiety. The reaction proceeds efficiently at mild temperatures, typically between -10°C and 25°C, ensuring thermal stability throughout the process.
![Final cyclization step to form 5-benzyl-furo[3,2-c]piperidine hydrochloride](/insights/img/furo-piperidine-synthesis-pharma-supplier-20260308235324-09.webp)
Impurity control is another critical aspect managed by the specific reaction conditions outlined in the patent. The use of formalin as the source of the one-carbon unit for the piperidine ring ensures precise stoichiometry, reducing the formation of oligomeric byproducts. Furthermore, the subsequent acidification step converts the free base into a stable salt, which precipitates out of the organic solvent mixture. This precipitation serves as an in-situ purification step, effectively excluding non-basic impurities and unreacted starting materials from the final isolate. For quality control teams, this mechanism provides a robust handle on the impurity profile, ensuring that the final API intermediate meets stringent regulatory specifications without the need for extensive rework.
How to Synthesize 5-Benzyl-Furo[3,2-c]Piperidine Efficiently
The synthesis of the core furo-piperidine scaffold is achieved through a logical sequence of four distinct chemical transformations. Beginning with the Henry reaction to install the nitrogen handle, followed by reduction to the amine, condensation with benzaldehyde, and finally the cyclization, each step is optimized for high yield and purity. The detailed standardized operating procedures for each stage, including specific temperature controls, addition rates, and workup protocols, are essential for reproducing the results on a pilot or commercial scale. Operators must pay close attention to the exothermic nature of the initial condensation and the moisture sensitivity of the reduction steps to ensure safety and consistency.
- Condense furfural with nitromethane using a mineral alkali catalyst to form 2-nitrovinylfuran.
- Reduce the nitro group to an amine using lithium aluminum hydride or catalytic hydrogenation to obtain furan ethylamine.
- Perform condensation with an aldehyde followed by reduction to form the secondary amine intermediate.
- Execute the final Pictet-Spengler cyclization using formaldehyde and acid to close the piperidine ring.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this new synthetic route offers tangible benefits that extend beyond simple chemistry. The primary driver of value is the substitution of high-cost, low-availability starting materials with commodity chemicals. Furfural is produced globally on a massive scale from agricultural waste, ensuring a stable and resilient supply chain that is less susceptible to market volatility. This raw material security translates directly into predictable pricing and reliable delivery schedules for downstream customers. Additionally, the simplification of the purification workflow removes the dependency on specialized chromatography resins and the associated disposal costs, further enhancing the economic viability of the project.
- Cost Reduction in Manufacturing: The elimination of expensive 3-furfural and the removal of column chromatography steps result in substantial cost savings. By relying on crystallization and extraction, the process reduces solvent usage and labor hours significantly. The use of catalytic hydrogenation or inexpensive hydride reagents for reduction steps further optimizes the reagent cost profile, making the final intermediate highly competitive in the global market.
- Enhanced Supply Chain Reliability: Sourcing furfural and nitromethane is straightforward, as these are bulk chemicals with multiple qualified suppliers worldwide. This diversification of the supply base mitigates the risk of single-source failures. Furthermore, the robustness of the reaction conditions means that the process is less sensitive to minor variations in raw material quality, reducing the rejection rate of incoming shipments and ensuring continuous production flow.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from gram-scale laboratory experiments to ton-scale commercial production. The avoidance of heavy metal catalysts in the main synthetic line simplifies waste treatment and reduces the environmental footprint. The ability to isolate the product as a salt directly from the reaction mixture minimizes the generation of aqueous waste streams, aligning with strict environmental regulations and sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and process descriptions provided in the patent documentation. Understanding these nuances is vital for technical teams evaluating the feasibility of adopting this route for their specific pipeline projects. The answers highlight the balance between chemical efficiency and practical manufacturability.
Q: What are the key advantages of using furfural over 3-furfural in this synthesis?
A: Furfural is significantly cheaper and more readily available than 3-furfural. Additionally, the new process avoids complex column purification steps required by older methods, drastically simplifying downstream processing.
Q: How does the Pictet-Spengler reaction contribute to the structural integrity of the final product?
A: The acid-catalyzed Pictet-Spengler cyclization efficiently closes the piperidine ring onto the furan scaffold, creating the rigid furo[3,2-c]piperidine core essential for JAK kinase inhibitory activity.
Q: Is this synthetic route suitable for large-scale commercial production?
A: Yes, the route is designed for scalability. It utilizes common solvents like methanol and toluene, avoids expensive transition metal catalysts in the early steps, and relies on crystallization rather than chromatography for purification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Furo-Piperidine Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient process chemistry plays in accelerating drug development timelines. Our team of expert process chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab to plant is seamless. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our capability to handle complex heterocyclic syntheses, such as the furo-piperidine scaffold, positions us as a strategic partner for your long-term supply needs.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific requirements. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits applicable to your project volume. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data and proven expertise. Let us collaborate to bring your next-generation kinase inhibitors to market faster and more cost-effectively.