Advanced Synthesis of 17-Hydroxycapsaicin for Scalable Pharmaceutical Intermediate Production
The pharmaceutical industry's relentless pursuit of understanding drug metabolism has placed a premium on high-purity metabolite standards, particularly for widely used bioactive compounds like capsaicin. Patent CN115850108A introduces a groundbreaking synthetic methodology for 17-hydroxycapsaicin, a critical omega-1 metabolite that plays a pivotal role in pharmacokinetic profiling and safety evaluation. This innovative approach addresses the longstanding scarcity of reliable reference standards by offering a streamlined, high-efficiency route that bypasses the limitations of traditional extraction or convoluted synthetic pathways. By leveraging a strategic combination of olefin metathesis and mild amidation conditions, the technology ensures the production of material with exceptional purity, directly supporting rigorous in vivo studies and regulatory submissions. For R&D directors and procurement specialists, this patent represents a significant leap forward in securing a stable supply chain for complex pharmaceutical intermediates, enabling more accurate toxicity assessments and metabolic mapping without the bottlenecks of legacy methods.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of specific capsaicin metabolites like 17-hydroxycapsaicin has been fraught with significant technical and economic challenges that hinder large-scale research and development efforts. Traditional methods often rely on the isolation of trace metabolites from biological matrices, a process that is not only labor-intensive and time-consuming but also yields insufficient quantities for comprehensive toxicological screening. Alternatively, existing synthetic routes frequently involve harsh reaction conditions, multiple protection-deprotection steps, and the use of expensive or hazardous reagents that complicate purification and drive up costs. These inefficiencies result in low overall yields and inconsistent purity profiles, which can introduce variability into analytical data and compromise the validity of pharmacokinetic studies. Furthermore, the lack of a scalable, robust manufacturing process has created a supply bottleneck, forcing research teams to delay critical projects or rely on suboptimal substitutes that do not accurately reflect the metabolic behavior of the parent drug in human systems.
The Novel Approach
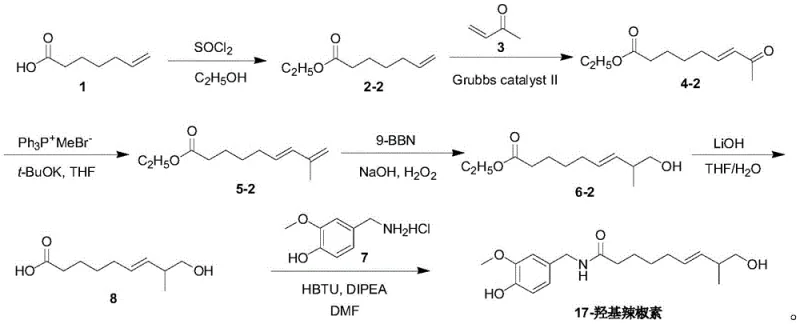
In stark contrast to these legacy challenges, the methodology disclosed in CN115850108A presents a rationally designed, convergent synthesis that prioritizes operational simplicity and chemical efficiency from the outset. The core innovation lies in the construction of the fatty acid side chain through a sequence of esterification, olefin metathesis, and regioselective hydroboration, which allows for precise control over the position of the hydroxyl group at the C17 position. This route eliminates the need for complex chiral resolutions or extreme temperatures, utilizing readily available starting materials such as 6-heptenoic acid to build the carbon skeleton with high atom economy. The final assembly of the molecule is achieved through a highly efficient amidation reaction under mild conditions, ensuring the integrity of the sensitive vanillyl moiety is preserved throughout the process. This streamlined approach not only drastically simplifies the workflow but also enhances the reproducibility of the synthesis, making it an ideal candidate for reliable pharmaceutical intermediate supplier operations seeking to deliver consistent quality at scale.
Mechanistic Insights into HBTU-Mediated Amidation and Hydroboration
The chemical elegance of this synthesis is perhaps best exemplified by the final amidation step, which utilizes HBTU (benzotriazole-N,N,N',N'-tetramethyluronium hexafluorophosphate) as a coupling reagent to join the functionalized fatty acid with the vanillylamine derivative. This mechanism proceeds through the formation of an active ester intermediate, which significantly lowers the activation energy required for amide bond formation while minimizing the risk of racemization or side reactions that are common with carbodiimide-based reagents. The reaction is conducted at controlled temperatures between 0°C and 30°C in the presence of diisopropylethylamine, a non-nucleophilic base that scavenges protons without interfering with the electrophilic centers. This precise control over reaction kinetics ensures that the phenolic hydroxyl groups on the aromatic ring remain unprotected yet unreactive, thereby eliminating the need for additional protection steps that would otherwise add cost and complexity to the manufacturing process. The result is a clean conversion to the target amide with minimal byproduct formation, facilitating straightforward downstream purification via column chromatography or crystallization.
Complementing the final coupling is the strategic use of hydroboration-oxidation to install the critical hydroxyl functionality with high regioselectivity. By employing 9-borabicyclo[3.3.1]nonane (9-BBN), the synthesis achieves anti-Markovnikov addition across the terminal alkene, ensuring that the oxygen atom is placed precisely at the omega-1 position rather than the terminal carbon. This selectivity is paramount for generating the correct metabolite structure, as even minor isomeric impurities could skew metabolic data. The subsequent oxidation with alkaline hydrogen peroxide proceeds smoothly under mild thermal conditions, preserving the stereochemistry of the adjacent double bond and preventing over-oxidation of the sensitive allylic system. This mechanistic precision underscores the route's suitability for producing high-purity capsaicin metabolite standards, as it inherently suppresses the formation of structural analogues that are difficult to separate and could interfere with analytical quantification in biological samples.
How to Synthesize 17-Hydroxycapsaicin Efficiently
Implementing this synthesis in a production environment requires a disciplined approach to process control, particularly regarding the management of exothermic reactions and the purification of intermediates. The protocol begins with the esterification of 6-heptenoic acid, followed by chain extension via olefin metathesis, which must be monitored closely to ensure complete conversion before proceeding to the Wittig olefination. Each step builds upon the previous one to construct the complex lipid tail, culminating in the hydrolysis of the ester to reveal the free carboxylic acid necessary for the final coupling. The detailed standardized synthetic steps see the guide below, which outlines the specific molar ratios, solvent systems, and workup procedures required to achieve the reported yields and purity levels consistently. Adhering to these parameters is essential for maintaining the integrity of the supply chain and ensuring that the final product meets the stringent specifications required for regulatory-grade reference materials.
- Perform esterification of 6-heptenoic acid followed by olefin metathesis with methyl vinyl ketone using Grubbs catalyst to extend the carbon chain.
- Execute Wittig reaction to introduce terminal alkene, followed by regioselective hydroboration-oxidation using 9-BBN to install the hydroxyl group.
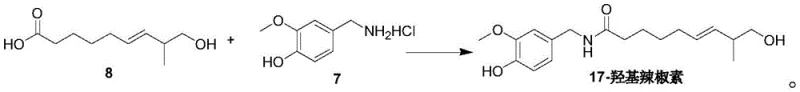
- Conduct ester hydrolysis to obtain the acid intermediate, then couple with vanillylamine derivative via HBTU-mediated amidation to finalize the target molecule.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers profound advantages that directly address the pain points of procurement managers and supply chain heads responsible for sourcing complex fine chemicals. The reliance on commodity chemicals such as 6-heptenoic acid and methanol significantly reduces raw material costs and mitigates the risk of supply disruptions associated with exotic or proprietary reagents. Furthermore, the elimination of transition metal catalysts in the final steps and the use of mild reaction conditions translate to lower energy consumption and reduced waste generation, aligning with modern sustainability goals and environmental compliance standards. The robustness of the process allows for seamless scale-up from laboratory benchtop to industrial reactor volumes without the need for specialized high-pressure or cryogenic equipment, thereby enhancing supply chain reliability and reducing lead time for high-purity capsaicin metabolites. These factors combine to create a cost-effective manufacturing model that ensures long-term availability of this critical intermediate for the global pharmaceutical market.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by utilizing readily available starting materials and avoiding expensive chiral catalysts or protecting group strategies that typically inflate production budgets. The high atom economy of the olefin metathesis and hydroboration steps minimizes raw material waste, while the efficient amidation protocol reduces the consumption of coupling reagents and solvents. Additionally, the simplified purification workflow lowers the operational overhead associated with chromatography and recrystallization, resulting in substantial cost savings that can be passed down the supply chain. This economic efficiency makes the commercial scale-up of complex pharmaceutical intermediates financially viable even in a competitive market environment.
- Enhanced Supply Chain Reliability: By sourcing raw materials that are commercially abundant and chemically stable, the synthesis mitigates the risks associated with single-source suppliers or volatile market pricing for specialty reagents. The operational simplicity of the reaction steps means that production can be easily transferred between manufacturing sites or scaled up rapidly to meet surges in demand without compromising quality. This flexibility ensures a continuous supply of 17-hydroxycapsaicin, preventing project delays for downstream drug development teams who rely on timely delivery of metabolite standards for their toxicology and PK studies. The robustness of the route effectively decouples production capacity from the constraints of complex synthetic logistics.
- Scalability and Environmental Compliance: The synthetic design inherently supports green chemistry principles by operating at near-ambient temperatures and generating minimal hazardous byproducts, which simplifies waste treatment and disposal procedures. The absence of heavy metal residues in the final product reduces the burden on quality control laboratories to perform extensive cleansing steps, accelerating the release of batches for commercial distribution. This environmental compatibility not only lowers regulatory compliance costs but also enhances the corporate sustainability profile of the manufacturer. The process is engineered for scalability, allowing for the transition from gram-scale research samples to multi-kilogram commercial production with consistent quality and minimal process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 17-hydroxycapsaicin synthesized via this patented method. These answers are derived directly from the technical specifications and beneficial effects outlined in the patent documentation, providing clarity on purity, scalability, and isotopic labeling capabilities. Understanding these aspects is crucial for stakeholders evaluating the feasibility of integrating this intermediate into their research pipelines or supply chains. The responses highlight the method's versatility and its alignment with industry standards for pharmaceutical reference materials.
Q: How does this synthesis route control impurity profiles for pharmacokinetic studies?
A: The route utilizes mild reaction conditions such as low-temperature amidation and selective hydroboration, which minimize side reactions and byproduct formation, ensuring high chemical purity essential for metabolic standard references.
Q: Is the process scalable for commercial supply of capsaicin metabolites?
A: Yes, the synthesis relies on readily available starting materials like 6-heptenoic acid and employs robust catalytic steps like olefin metathesis, which are well-established for kilogram-to-ton scale manufacturing without complex equipment requirements.
Q: Can this method be adapted for isotope-labeled analogues?
A: The modular nature of the synthesis, particularly the final amidation step, allows for the straightforward substitution of the amine component with deuterated or C13-labeled vanillylamine derivatives to produce tracers for ADME studies.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 17-Hydroxycapsaicin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having access to high-quality metabolite standards for advancing drug discovery and development programs. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of 17-hydroxycapsaicin delivered meets the highest industry standards for analytical accuracy. We are committed to supporting your research goals by providing a stable, scalable source of this valuable intermediate, allowing your team to focus on innovation rather than supply chain uncertainties.
We invite you to engage with our technical procurement team to discuss how our manufacturing capabilities can optimize your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain insights into how our efficient synthesis route can reduce your overall material costs while improving lead times. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your volume and purity needs. Let us partner with you to ensure the success of your pharmacokinetic studies and regulatory submissions through our dedicated supply of premium pharmaceutical intermediates.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →