Advanced Synthetic Route for Substituted Isoindolines: Overcoming Steric Hindrance for Commercial Scale-Up
Advanced Synthetic Route for Substituted Isoindolines: Overcoming Steric Hindrance for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust and scalable methodologies for constructing complex heterocyclic scaffolds, particularly isoindoline derivatives which serve as critical building blocks in numerous drug candidates. Patent CN112028816A, published in December 2020, introduces a transformative synthetic strategy that addresses long-standing challenges in introducing sterically demanding substituents onto the isoindoline core. Unlike traditional approaches that struggle with bulky groups, this invention leverages a clever ring-opening and reclosing mechanism mediated by organometallic reagents. This technical breakthrough not only expands the chemical space accessible to medicinal chemists but also offers significant implications for process chemistry and supply chain stability. By shifting from direct alkylation to a nucleophilic addition-reduction-cyclization sequence, the method ensures high purity and operational simplicity, making it an attractive option for reliable pharmaceutical intermediate supplier networks aiming to diversify their portfolio of complex heterocycles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of substituted isoindolines has been plagued by significant structural limitations, primarily dictated by the reliance on strong base-mediated alkylation strategies. As highlighted in the background art of the patent, prior methods such as those disclosed by AbbVie utilize powerful bases like lithium hexamethyldisilazide (LHMDS) to deprotonate the methylene position of Boc-protected isoindolinones. While effective for introducing small, highly reactive alkyl groups like methyl or benzyl, this approach fundamentally fails when attempting to incorporate bulkier or less reactive functionalities. The steric congestion around the nitrogen atom prevents the successful displacement of leaving groups by larger nucleophiles, effectively capping the diversity of derivatives that can be produced. Furthermore, the harsh conditions required for these strong base reactions often lead to side reactions and decomposition, complicating the purification process and reducing overall yield, which is a critical bottleneck for cost reduction in API manufacturing.
The Novel Approach
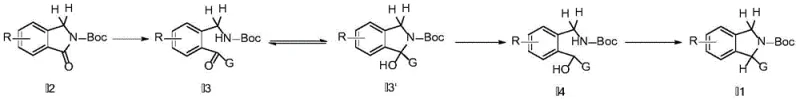
The methodology presented in CN112028816A circumvents these steric barriers through an innovative ring-opening strategy that fundamentally alters the reaction trajectory. Instead of attempting to force a bulky group onto the nitrogen-adjacent carbon directly, the process initiates with a nucleophilic attack by a Grignard or lithium reagent on the carbonyl carbon of the lactam ring. This attack temporarily opens the five-membered ring, generating a ketone or hemiacetal intermediate where the new substituent is attached to what was originally the carbonyl carbon. This open-chain configuration eliminates the immediate steric clash associated with the closed ring system, allowing even bulky groups like isopropyl, phenyl, or heteroaryl rings to be installed with high efficiency. Subsequent reduction and activation steps then facilitate the reconstruction of the isoindoline core, locking the bulky substituent into the desired 1-position with remarkable precision and stability.
Mechanistic Insights into Grignard-Mediated Ring Opening and Cyclization
The core of this synthetic innovation lies in the precise control of organometallic addition and subsequent functional group transformations. The reaction begins with the treatment of Boc-protected isoindolinone with a Grignard or lithium reagent at cryogenic temperatures, typically ranging from -40°C to -78°C. At this stage, the nucleophile attacks the electrophilic carbonyl carbon, breaking the amide bond and forming a tetrahedral intermediate that collapses into a ketone (when G is aryl/heteroaryl) or exists in equilibrium with a hemiacetal (when G is alkyl). This step is critical because it converts a rigid cyclic system into a flexible acyclic precursor, thereby relieving the torsional strain that usually inhibits the introduction of large groups. The use of aprotic solvents like tetrahydrofuran ensures the stability of the organometallic species, while the low temperature suppresses potential side reactions such as over-addition or polymerization, ensuring a clean conversion to the key intermediate.
Following the formation of the ketone or hemiacetal, the pathway proceeds through a reduction step using mild hydride sources such as sodium borohydride. This reduction converts the carbonyl functionality into a secondary alcohol, setting the stage for the final ring-closing event. The hydroxyl group is then activated using sulfonyl chlorides like methanesulfonyl chloride (MsCl) or p-toluenesulfonyl chloride (TsCl) in the presence of a mild base such as triethylamine. This activation transforms the poor leaving hydroxyl group into an excellent leaving group (mesylate or tosylate). Under these mildly basic conditions, the pendant amine nitrogen performs an intramolecular nucleophilic substitution, displacing the sulfonate and reforming the five-membered isoindoline ring. This intramolecular cyclization is entropically favored and proceeds smoothly even with the newly installed bulky substituents, delivering the final high-purity isoindoline derivatives with excellent regioselectivity.
How to Synthesize Substituted Isoindoline Efficiently
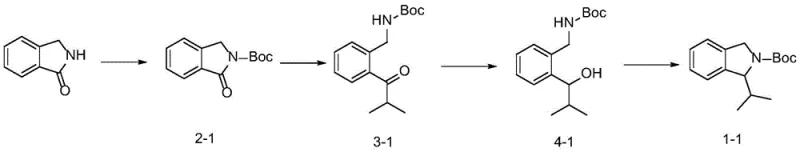
The practical implementation of this synthesis involves a streamlined three-step sequence that is highly amenable to standard laboratory and pilot plant equipment. The process begins with the preparation of the starting material, Boc-protected isoindolinone, which is readily accessible and stable. The subsequent addition of the Grignard reagent requires careful temperature control but utilizes common reagents found in any fine chemical facility. Following the workup, the crude ketone intermediate can often be carried forward without extensive purification, further simplifying the workflow. The reduction and cyclization steps are equally straightforward, employing reagents that are inexpensive and easy to handle on a large scale. For detailed operational parameters, stoichiometry, and specific workup procedures tailored to different substituents, refer to the standardized synthesis guide below.
- React Boc-protected isoindolinone with a Grignard or lithium reagent at low temperatures (-78°C) to form a ketone or hemiacetal intermediate.
- Reduce the resulting ketone or hemiacetal intermediate using a hydride reducing agent like sodium borohydride to obtain the corresponding alcohol.
- Activate the hydroxyl group with a sulfonyl chloride (e.g., MsCl or TsCl) and induce cyclization under mild basic conditions to form the final substituted isoindoline.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this synthetic route offers compelling advantages that directly address the pain points of sourcing complex heterocyclic intermediates. The elimination of exotic catalysts and the reliance on commodity chemicals like Grignard reagents and sodium borohydride significantly de-risks the supply chain. Traditional methods often depend on specialized reagents or conditions that can be difficult to source consistently in multi-ton quantities, leading to potential bottlenecks. In contrast, the reagents used in this patent are staples of the chemical industry, ensuring a stable and continuous supply flow. Moreover, the operational simplicity reduces the need for highly specialized equipment, allowing for production in a wider range of manufacturing facilities, which enhances supply chain resilience and reduces lead time for high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the simplification of the synthetic route and the avoidance of expensive materials. By bypassing the need for precious metal catalysts or complex chiral ligands often required in alternative asymmetric syntheses, the raw material costs are drastically lowered. Additionally, the high yields reported in the examples, combined with the ability to telescope steps or minimize purification requirements, result in substantial cost savings per kilogram of product. The mild reaction conditions also translate to lower energy consumption for heating or cooling compared to processes requiring extreme temperatures or pressures, contributing to a more sustainable and cost-effective manufacturing profile.
- Enhanced Supply Chain Reliability: The robustness of this chemistry ensures consistent quality and availability, which is paramount for long-term drug development projects. Because the method tolerates a wide variety of functional groups and substituents, it provides a versatile platform technology that can be adapted to produce a library of analogues without re-validating entirely new processes. This flexibility means that if a specific analogue is needed, the supply chain can respond rapidly without the long lead times associated with developing bespoke synthetic routes from scratch. The use of stable intermediates also allows for strategic stockpiling, further buffering against market volatility and ensuring uninterrupted delivery to downstream customers.
- Scalability and Environmental Compliance: Scaling this process from gram to ton scale is facilitated by the absence of hazardous or difficult-to-handle reagents. The waste streams generated are primarily organic salts and solvents which are well-understood and manageable within standard wastewater treatment protocols, unlike processes generating heavy metal waste. The high atom economy of the cyclization step, where the majority of the molecular framework is retained in the final product, minimizes waste generation. This alignment with green chemistry principles not only reduces disposal costs but also ensures compliance with increasingly stringent environmental regulations, making it a future-proof choice for commercial scale-up of complex heterocycles.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic technology. These answers are derived directly from the experimental data and claims within the patent documentation, providing a factual basis for decision-making. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this route into their existing manufacturing pipelines.
Q: What are the limitations of conventional isoindoline synthesis methods?
A: Conventional methods often rely on strong bases like LHMDS for direct alkylation, which fail to introduce bulky or weakly reactive groups such as phenyl or isopropyl due to steric hindrance and low reactivity.
Q: How does the new synthetic method overcome steric hindrance?
A: The novel approach utilizes a ring-opening strategy where a Grignard reagent attacks the carbonyl carbon, temporarily breaking the ring structure. This bypasses the steric constraints of direct N-alkylation, allowing for the introduction of bulky substituents before reclosing the ring.
Q: Is this process suitable for large-scale pharmaceutical manufacturing?
A: Yes, the method employs readily available reagents like Grignard reagents and sodium borohydride under relatively mild conditions, avoiding expensive transition metal catalysts and complex purification steps, which facilitates commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Substituted Isoindoline Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug discovery and development. Our team of expert process chemists has thoroughly analyzed the technology disclosed in CN112028816A and is fully equipped to translate this laboratory-scale innovation into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from clinical trials to market launch. Our state-of-the-art facilities are designed to handle sensitive organometallic reactions with precision, and our rigorous QC labs enforce stringent purity specifications to guarantee that every batch meets the highest international standards.
We invite you to leverage our technical expertise to optimize your supply chain for isoindoline derivatives. Whether you require custom synthesis of specific analogues or large-scale production of the core scaffold, our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to innovation and quality can drive value for your organization.