Advanced Rhodium-Catalyzed Synthesis of Terminal Alkenyl Indole Derivatives for Commercial Scale-Up
Introduction to Patent CN115650953A Technology
The landscape of fine organic chemical synthesis is constantly evolving, driven by the need for more efficient routes to complex heterocyclic scaffolds essential for modern drug discovery. Patent CN115650953A introduces a groundbreaking methodology for the preparation of terminal alkenyl indole derivatives, specifically addressing the long-standing challenge of functionalizing the C7 position of the indole ring. Historically, the inherent reactivity of the C2 and C3 positions has made direct C7 modification exceptionally difficult, often requiring harsh conditions or multi-step protection-deprotection sequences that degrade overall process efficiency. This patent discloses a robust catalytic system that overcomes these kinetic barriers, enabling the direct installation of terminal alkenyl groups with high precision.
The core innovation lies in the strategic use of an N-pyridyl directing group which coordinates with a rhodium catalyst to activate the proximal C7-H bond. This approach not only solves the regioselectivity issue but also ensures that the resulting terminal olefin structure remains intact for downstream transformations. For R&D directors and process chemists, this represents a significant leap forward in accessing diverse chemical space for structure-activity relationship (SAR) studies. The ability to introduce varied aryl and alkyl groups at the C7 position opens new avenues for developing bioactive molecules where the indole core is a privileged structure.
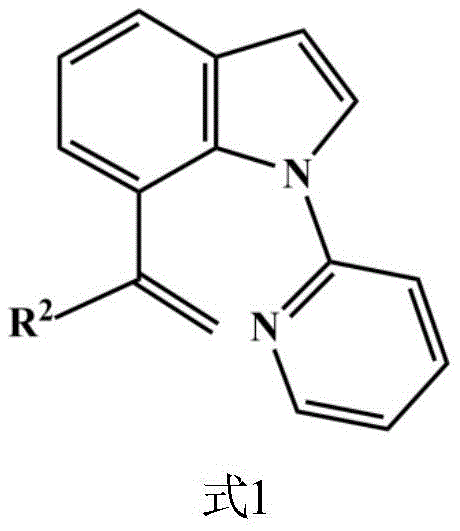
Furthermore, the practical implications of this technology extend beyond the laboratory bench. By establishing a reliable pathway to these specific derivatives, the patent lays the groundwork for scalable manufacturing processes that can meet the rigorous demands of the pharmaceutical industry. The described compounds, characterized by their terminal alkenyl motifs, serve as versatile building blocks for further functionalization, such as cross-coupling reactions or cycloadditions. As a reliable pharmaceutical intermediates supplier, understanding and leveraging such patented methodologies is crucial for maintaining a competitive edge in the global supply chain of high-value fine chemicals.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional strategies for synthesizing alkenyl indoles have frequently encountered substantial hurdles, particularly when targeting the C7 position. Conventional electrophilic substitution reactions typically favor the electron-rich C3 position, making C7 functionalization nearly impossible without extensive structural modification of the indole nucleus. Existing methods often rely on pre-functionalized starting materials, such as C7-halogenated indoles, which are themselves expensive and difficult to procure in bulk quantities. Moreover, when non-terminal alkenyl structures are formed, the resulting internal olefins lack the synthetic utility of terminal alkenes, limiting their application in subsequent derivatization steps required for complex molecule assembly.
Another critical drawback of prior art is the instability of the olefinic moiety under the vigorous conditions often required for C-H activation. High temperatures and strong oxidants can lead to polymerization or isomerization of the double bond, resulting in complex mixtures that are difficult to purify. This not only reduces the overall yield but also complicates the impurity profile, posing significant risks for regulatory compliance in pharmaceutical manufacturing. The inability to consistently produce high-purity terminal alkenyl indoles has thus remained a bottleneck, restricting the exploration of this chemical class in medicinal chemistry programs and delaying the development of potential new therapeutics.
The Novel Approach
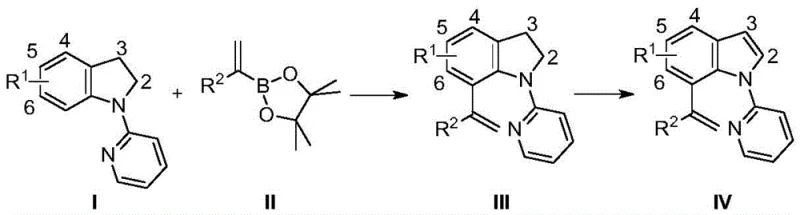
The methodology outlined in CN115650953A offers a transformative solution by employing a Rhodium(III)-catalyzed C-H activation strategy coupled with a mild oxidative workup. This novel approach utilizes readily available N-pyridylindoline derivatives as substrates, which act as effective templates for regioselective C-H bond cleavage. The reaction proceeds under relatively mild conditions, typically around 60°C in methanol, utilizing alkenyl boronates as the coupling partners. This choice of reagents is significant because boronates are generally stable, commercially available, and tolerate a wide range of functional groups, thereby enhancing the versatility of the synthesis.
Crucially, the process is divided into two distinct stages: the initial alkenylation to form an indoline intermediate, followed by a separate oxidative aromatization step using manganese dioxide. This separation of concerns allows for optimal control over each transformation. The first step installs the carbon framework with high fidelity, while the second step gently restores aromaticity without compromising the sensitive terminal double bond. This strategic decoupling ensures that the final products, such as the 7-styryl-N-pyridylindole derivatives, are obtained with exceptional purity and yields often exceeding 90 percent. Such efficiency is paramount for cost reduction in fine chemical manufacturing, as it minimizes waste generation and maximizes the throughput of valuable intermediates.
Mechanistic Insights into Rh(III)-Catalyzed C-H Alkenylation
To fully appreciate the robustness of this synthesis, one must delve into the mechanistic underpinnings of the Rhodium(III) catalytic cycle. The reaction initiates with the coordination of the pyridine nitrogen atom to the cationic Rh(III) species generated in situ from the dimeric precursor [RhCp*Cl2]2 and the silver salt additive. This coordination directs the metal center to the adjacent C7 position of the indoline ring, facilitating the cleavage of the C-H bond through a concerted metalation-deprotonation (CMD) pathway. This step is the turnover-limiting factor in many C-H activation processes, but the presence of the directing group significantly lowers the activation energy, allowing the reaction to proceed efficiently at moderate temperatures.
Following C-H activation, the resulting organorhodium intermediate undergoes transmetallation with the alkenyl boronate ester. This step transfers the alkenyl group to the metal center, setting the stage for the formation of the new carbon-carbon bond. Subsequent reductive elimination releases the C7-alkenylated indoline product and regenerates the active Rh(I) species, which is then re-oxidized by the silver carbonate to close the catalytic cycle. The second phase of the synthesis involves the treatment of this saturated indoline intermediate with manganese dioxide. This oxidant selectively dehydrogenates the five-membered ring, restoring the aromatic indole system while leaving the exocyclic terminal alkene untouched. This chemoselectivity is a hallmark of the process, ensuring that the desired terminal olefin functionality is preserved for future synthetic manipulations.
From an impurity control perspective, this mechanism offers distinct advantages. The high regioselectivity imposed by the directing group minimizes the formation of C2 or C3 isomers, which are common byproducts in non-directed indole functionalizations. Furthermore, the use of manganese dioxide for aromatization is a clean transformation that produces insoluble manganese byproducts, which can be easily removed by simple filtration. This simplifies the downstream purification process, reducing the burden on chromatographic separation and lowering the overall solvent consumption. For quality control teams, this translates to a cleaner crude product profile and a more straightforward path to meeting stringent purity specifications required for GMP manufacturing environments.
How to Synthesize Terminal Alkenyl Indole Derivatives Efficiently
Implementing this synthesis in a production setting requires careful attention to reaction parameters and stoichiometry to ensure reproducibility and safety. The patent provides a detailed blueprint for executing the two-step sequence, emphasizing the importance of maintaining anhydrous conditions during the catalytic step and controlling the temperature during the oxidation phase. The molar ratio of the N-pyridylindoline to the alkenyl boronate is typically optimized between 1:1 and 1:4, with a preference for a slight excess of the boronate to drive the reaction to completion. The catalyst loading is kept low, often around 2 mol%, which is economically favorable for large-scale operations.
- Mix N-pyridylindoline derivatives with alkenyl boronates in the presence of [RhCp*Cl2]2 catalyst and Ag2CO3 oxidant in methanol at 60°C.
- Isolate the intermediate indoline product via silica gel column chromatography.
- Oxidize the intermediate using Manganese Dioxide (MnO2) in 1,2-dichloroethane at reflux (≥85°C) to obtain the final terminal alkenyl indole.
Following the initial coupling, the workup involves standard extraction and concentration techniques, but the key purification step utilizes silica gel column chromatography with a petroleum ether and ethyl acetate gradient. This allows for the isolation of the intermediate indoline in high purity before it is subjected to the oxidation step. The oxidation itself is performed in 1,2-dichloroethane at reflux temperatures (≥85°C), ensuring complete conversion to the aromatic indole. The final product is then isolated via filtration to remove the spent oxidant and further purified if necessary. This standardized protocol ensures that the process can be reliably scaled from gram to kilogram quantities without loss of efficiency.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits that directly impact the bottom line and operational resilience. The primary advantage lies in the accessibility of the starting materials. N-pyridylindolines and alkenyl boronates are commodity chemicals that can be sourced from multiple vendors globally, reducing the risk of supply chain disruptions associated with proprietary or exotic reagents. This diversification of the supply base enhances negotiation leverage and stabilizes pricing, contributing to significant cost reduction in fine chemical manufacturing. Additionally, the high atom economy of the C-H activation step means that less raw material is wasted, further optimizing the cost structure of the final product.
Operational efficiency is another critical factor. The reaction conditions are relatively mild, avoiding the need for specialized high-pressure equipment or cryogenic cooling systems that drive up capital expenditure. The use of common solvents like methanol and 1,2-dichloroethane simplifies solvent recovery and recycling protocols, aligning with modern sustainability goals. Moreover, the high yields reported in the patent, often surpassing 90 percent for the final products, imply that less capacity is tied up in reprocessing off-spec material. This increased throughput allows manufacturers to respond more quickly to market demand, effectively reducing lead time for high-purity pharmaceutical intermediates and ensuring a steady flow of materials to downstream customers.
- Cost Reduction in Manufacturing: The elimination of expensive pre-functionalized starting materials and the use of low catalyst loadings significantly lower the bill of materials. The streamlined two-step process reduces labor hours and utility consumption compared to multi-step traditional routes, driving down the overall cost of goods sold without compromising quality.
- Enhanced Supply Chain Reliability: By relying on widely available boronate esters and simple indoline precursors, the risk of single-source dependency is mitigated. The robustness of the reaction against various functional groups means that supply fluctuations for specific substituted variants can be managed by switching to alternative substrates within the same chemical family, ensuring continuous production.
- Scalability and Environmental Compliance: The process generates minimal hazardous waste, primarily insoluble manganese salts which are easy to handle and dispose of according to environmental regulations. The high selectivity reduces the need for extensive chromatographic purification on a large scale, potentially allowing for crystallization-based purification which is more scalable and environmentally friendly.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a clear picture of what partners can expect when integrating this chemistry into their pipelines. Understanding these nuances is essential for making informed decisions about process adoption and resource allocation.
Q: What is the key advantage of this C7 functionalization method?
A: Unlike traditional methods that struggle with C7 selectivity due to C2/C3 reactivity, this Rh(III)-catalyzed protocol utilizes a pyridine directing group to achieve precise C7 alkenylation with high regioselectivity.
Q: Can this process accommodate diverse substrate scopes?
A: Yes, the patent demonstrates excellent tolerance for various substituents including halogens, alkyl groups, alkoxy groups, and electron-withdrawing groups on both the indole and the alkenyl boronate components.
Q: How is the terminal olefin stability maintained during synthesis?
A: The process employs a mild two-step strategy where the sensitive terminal olefin is installed on the indoline scaffold first, followed by a controlled oxidative aromatization that preserves the double bond integrity.
It is important to note that while the patent covers a broad scope of substrates, specific optimization may be required for highly sterically hindered or electronically deactivated variants. However, the fundamental robustness of the Rh-catalyzed system provides a strong foundation for troubleshooting and method development. Our technical team is equipped to assist clients in navigating these variables to ensure successful technology transfer and scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Terminal Alkenyl Indole Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of advanced synthetic methodologies like those described in CN115650953A. As a leading CDMO and supplier, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring these complex molecules to the market. Our facilities are equipped with state-of-the-art reactors capable of handling air-sensitive catalytic reactions and rigorous QC labs that ensure stringent purity specifications are met for every batch. We understand that consistency is key in the pharmaceutical supply chain, and our commitment to quality assurance guarantees that our terminal alkenyl indole derivatives perform reliably in your downstream applications.
We invite you to collaborate with us to leverage this cutting-edge chemistry for your drug discovery programs. Whether you require custom synthesis of specific analogs or bulk supply of standard intermediates, our team is ready to provide a Customized Cost-Saving Analysis tailored to your project needs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you accelerate your timeline and reduce costs by integrating this efficient synthesis into your supply chain.