Advanced Metal-Free Synthesis of Conjugated Alkenyl Amidines for Commercial Pharmaceutical Applications
Introduction to Next-Generation Amidine Synthesis
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for greener, more efficient, and cost-effective synthetic routes. A pivotal advancement in this domain is documented in Chinese Patent CN113135840A, which discloses a novel method for synthesizing conjugated alkenyl amidine compounds. These structures serve as critical building blocks for constructing nitrogen-containing heterocycles via Hetero-Diels-Alder strategies, essential for developing complex drug molecules including chiral piperidines and quinolines. Unlike traditional methodologies that rely heavily on transition metal catalysts, this innovative approach utilizes a fluoride-ion catalyzed one-pot reaction involving Kobayashi aryne precursor derivatives, N,N-dimethylformamide (DMF), and isonitrile compounds. This technological breakthrough not only streamlines the synthetic pathway but also addresses significant pain points regarding metal contamination and reaction severity, positioning it as a highly attractive option for reliable pharmaceutical intermediate supplier networks seeking to optimize their supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of conjugated alkenyl amidines has been fraught with challenges that hinder large-scale adoption. Early reports, such as the 1971 Saegusa methodology, relied on Copper(I) oxide (Cu2O) as a catalyst at elevated temperatures up to 80°C. While functional, this approach suffers from significant limitations, including incompatibility with saturated alkyl isonitrile compounds and unsubstituted amines, thereby restricting the chemical space accessible to medicinal chemists. Furthermore, the reliance on transition metals introduces a costly and time-consuming purification burden to remove trace metal residues, which is strictly regulated in API manufacturing. Subsequent attempts, such as the 1987 Marchesini method using Vilsmeier reagents, yielded disappointing results with conversion rates as low as 6%, rendering them economically unviable for industrial applications. Other methods involving bissilyl enamines demonstrated poor substrate universality, often failing to produce more than a couple of examples, which limits their utility in diverse drug discovery programs.
The Novel Approach
In stark contrast to these legacy techniques, the method outlined in CN113135840A represents a paradigm shift towards sustainable and efficient chemistry. By leveraging the unique reactivity of arynes generated in situ from Kobayashi precursors, this protocol enables the construction of the amidine backbone under exceptionally mild conditions. The reaction proceeds smoothly at room temperature (approximately 25°C) within a timeframe of just 5 hours, eliminating the need for energy-intensive heating. Crucially, the system operates without any transition metal or noble metal catalysts, utilizing instead a cost-effective fluoride salt system, specifically Potassium Fluoride complexed with 18-Crown-6. This metal-free nature drastically reduces the complexity of the workup procedure and ensures a cleaner impurity profile. The broad substrate tolerance allows for the incorporation of various functional groups, including halogens, alkyls, and alkoxy groups, making it a versatile tool for the commercial scale-up of complex pharmaceutical intermediates.
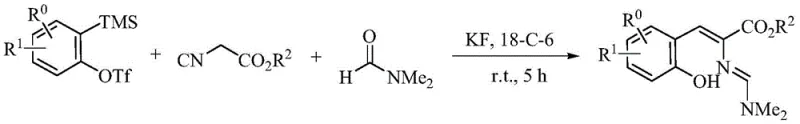
Mechanistic Insights into Fluoride-Catalyzed Aryne Cycloaddition
Understanding the underlying mechanism is vital for R&D directors aiming to replicate and optimize this process. The reaction initiates with the generation of a highly reactive benzyne intermediate from the Kobayashi precursor upon exposure to fluoride ions. This transient species immediately undergoes a [2+2] cycloaddition with the carbonyl group of the DMF solvent, forming a four-membered oxetane-like intermediate (Intermediate A). This unstable species rapidly isomerizes into an o-quinone methide derivative (Intermediate B), which acts as a potent electrophile. The isonitrile component then performs a nucleophilic attack on this intermediate, leading to the formation of Intermediate C. Through a series of intramolecular isomerizations and the elimination of dimethylamine, the structure rearranges into Intermediate D. Under the basic conditions provided by the fluoride source, deprotonation yields Intermediate E, which subsequently undergoes nucleophilic attack by dimethylamine on the isocyano-derived carbene carbon, followed by protonation to furnish the final stable conjugated alkenyl amidine product.
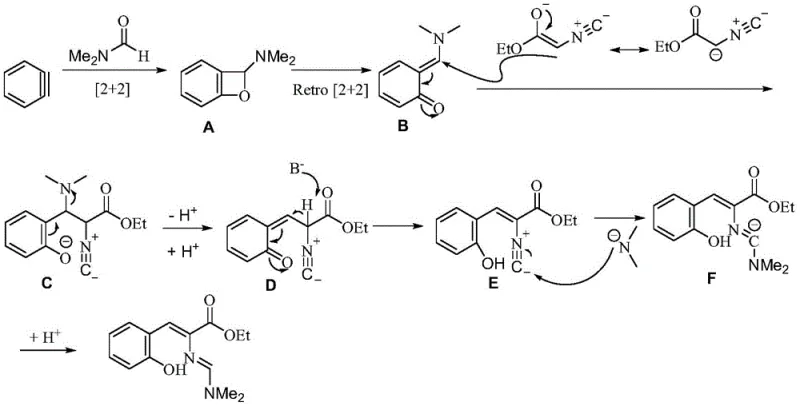
From an impurity control perspective, this mechanism offers distinct advantages over metal-catalyzed pathways. The absence of redox-active metal centers minimizes the risk of oxidative side reactions that often plague copper or palladium-catalyzed processes. Furthermore, the mild thermal conditions (25°C) suppress thermal decomposition pathways that could lead to polymeric byproducts or tar formation. The use of DMF as both solvent and reactant ensures a high local concentration of the carbonyl species, driving the initial cycloaddition forward efficiently and reducing the likelihood of benzyne dimerization or reaction with other nucleophiles present in the mixture. This inherent selectivity translates to a cleaner crude reaction mixture, facilitating easier isolation and higher overall purity of the final API intermediate, which is a critical metric for regulatory compliance.
How to Synthesize Conjugated Alkenyl Amidine Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific operational parameters to ensure reproducibility and safety. The protocol is designed as a straightforward one-pot procedure that minimizes unit operations. It begins with the preparation of the catalytic system, where precise stoichiometry of the fluoride source and phase transfer catalyst is essential to activate the aryne precursor effectively. The reaction is conducted under an inert nitrogen atmosphere to prevent moisture interference, which could quench the reactive benzyne species. Following the reaction period, a standard aqueous workup followed by chromatographic purification yields the target compound. For detailed operational specifics, the standardized synthesis steps are provided in the guide below.
- Prepare the reaction vessel by adding Potassium Fluoride (4.0 equiv) and 18-Crown-6 (1.0 equiv) to a dry Schlenk tube under nitrogen atmosphere.
- Add the isonitrile compound (1.0 equiv), Kobayashi aryne precursor derivative (1.5 equiv), and excess DMF solvent to the mixture.
- Stir the reaction at room temperature (25°C) for 5 hours, monitor by TLC, then quench with water, extract with ethyl acetate, and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this fluoride-catalyzed methodology offers tangible strategic benefits beyond mere chemical elegance. The primary advantage lies in the drastic simplification of the raw material portfolio. By eliminating the need for specialized transition metal catalysts, which are subject to volatile pricing and supply constraints, manufacturers can secure a more stable and predictable supply chain. Additionally, the removal of heavy metals from the process flow negates the requirement for expensive scavenging resins or complex extraction protocols typically mandated to meet strict residual metal specifications in pharmaceutical products. This streamlining directly contributes to substantial cost savings in API manufacturing by reducing both material costs and processing time.
- Cost Reduction in Manufacturing: The economic impact of switching to this metal-free protocol is profound. Traditional methods often incur hidden costs associated with the purchase of precious metal catalysts and the subsequent waste disposal fees for metal-contaminated solvents. By utilizing inexpensive commodity chemicals like Potassium Fluoride and DMF, the direct material cost is significantly lowered. Moreover, the simplified purification process reduces the consumption of silica gel and solvents during chromatography, further enhancing the overall cost efficiency. The ability to run the reaction at room temperature also eliminates energy costs associated with heating reactors, contributing to a leaner manufacturing budget without compromising on yield or quality.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of widely available, commodity-grade reagents. Unlike specialized organometallic catalysts that may have long lead times or single-source dependencies, potassium fluoride and crown ethers are standard inventory items for most chemical suppliers. This ubiquity ensures that production schedules are less likely to be disrupted by raw material shortages. Furthermore, the robustness of the reaction conditions means that the process is less sensitive to minor variations in reagent quality, allowing for greater flexibility in vendor selection and reducing the risk of batch failures due to supply chain inconsistencies.
- Scalability and Environmental Compliance: Scaling chemical processes often introduces new challenges, but this methodology is inherently scalable due to its mild exothermic profile and lack of hazardous metal waste. The absence of heavy metals simplifies environmental compliance, as wastewater treatment does not require specialized protocols for metal removal. This aligns with modern green chemistry principles and corporate sustainability goals, making the facility more attractive to eco-conscious partners. The high atom economy of the three-component reaction ensures that a majority of the starting materials end up in the final product, minimizing waste generation and maximizing resource efficiency in large-scale production environments.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating this technology for their specific applications, we have compiled answers to common inquiries regarding the reaction scope and optimization. These insights are derived directly from the experimental data presented in the patent documentation, ensuring accuracy and relevance for process development scientists. Understanding these nuances is key to successfully transferring this technology from the lab bench to commercial production.
Q: Why is the fluoride-catalyzed method superior to traditional transition metal catalysis for amidine synthesis?
A: Traditional methods often require expensive transition metals like Copper (Cu2O) which necessitate complex removal steps to meet pharmaceutical purity standards. The fluoride-catalyzed method described in CN113135840A operates under mild conditions without heavy metals, significantly simplifying downstream purification and reducing environmental impact.
Q: What is the optimal molar ratio of Kobayashi precursor to isonitrile for maximum yield?
A: Experimental data indicates that a molar ratio of 1.5:1 (Kobayashi precursor to isonitrile) provides the ideal balance, achieving yields above 85%. Deviating below a 1:1 ratio significantly drops the yield to below 56%.
Q: Can this synthesis be scaled up for industrial production?
A: Yes, the reaction utilizes a one-pot strategy at room temperature with commercially available reagents like DMF and KF. The absence of sensitive transition metal catalysts and the stability of the intermediates make it highly suitable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Conjugated Alkenyl Amidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced synthetic methodologies like the fluoride-catalyzed aryne reaction described in CN113135840A. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from discovery to market is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of conjugated alkenyl amidine intermediate adheres to the highest industry standards. We are committed to leveraging our technical expertise to optimize this metal-free route for your specific project needs, delivering high-quality intermediates that accelerate your drug development timeline.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits tailored to your volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that drive efficiency and profitability in your pharmaceutical manufacturing operations.