Scalable Production of 1-Carboxylic Acid Tert-Butyl Ester-3-Fluoro-Azetidine Derivatives
Scalable Production of 1-Carboxylic Acid Tert-Butyl Ester-3-Fluoro-Azetidine Derivatives
The pharmaceutical industry is constantly seeking more efficient pathways to synthesize complex heterocyclic building blocks, particularly fluorinated azetidines which are critical motifs in modern drug discovery. Patent CN102731362A introduces a groundbreaking methodology for preparing 1-carboxylic acid tert-butyl ester-3-fluoro-azetidine derivatives, addressing the severe inefficiencies of prior art. This technology represents a paradigm shift from laborious multi-step sequences to a concise, high-yielding three-step protocol. By leveraging electrophilic fluorination strategies and optimized protection group chemistry, this process delivers exceptional purity and operational simplicity. For R&D directors and procurement specialists alike, understanding this technological leap is essential for securing a competitive edge in the supply of high-value pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated azetidine scaffolds has been plagued by excessive step counts and prohibitive costs. As documented in literature such as J. Org. Chem. 74 (2009), traditional routes often necessitate up to eight distinct chemical transformations to reach the target molecule. These legacy processes rely heavily on stoichiometric amounts of expensive and toxic reagents, including palladium on carbon for hydrogenation and ruthenium trichloride for oxidation steps. The cumulative effect of such a long synthetic sequence results in a catastrophic loss of material, with reported total yields hovering around a mere 2.46%. Furthermore, the use of heavy metal catalysts introduces significant downstream purification challenges, requiring rigorous metal scavenging to meet stringent regulatory limits for active pharmaceutical ingredients. This not only inflates manufacturing costs but also extends lead times and complicates waste management protocols.
The Novel Approach
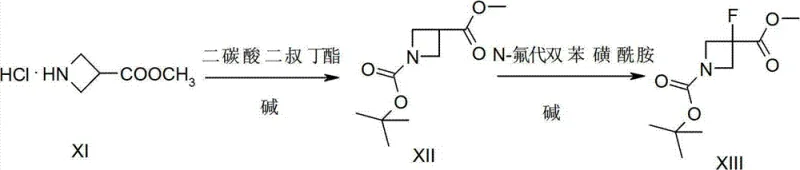
In stark contrast, the methodology disclosed in CN102731362A streamlines the entire synthesis into just three robust steps, dramatically improving efficiency and sustainability. The process initiates with the Boc-protection of methyl azetidine-3-carboxylate hydrochloride, followed by a direct electrophilic fluorination at the 3-position using N-fluorobenzenesulfonimide (NFSI). The final step involves a straightforward hydrolysis to yield the target carboxylic acid. This innovative route eliminates the need for transition metal catalysts entirely, replacing them with readily available organic reagents and bases. The result is a process that is not only operationally simpler but also economically superior, achieving a remarkable total yield of 74% for the intermediate and 99% for the final conversion. This drastic reduction in complexity translates directly into lower production costs and a more reliable supply chain for downstream API manufacturers.
Mechanistic Insights into Electrophilic Fluorination of Azetidines
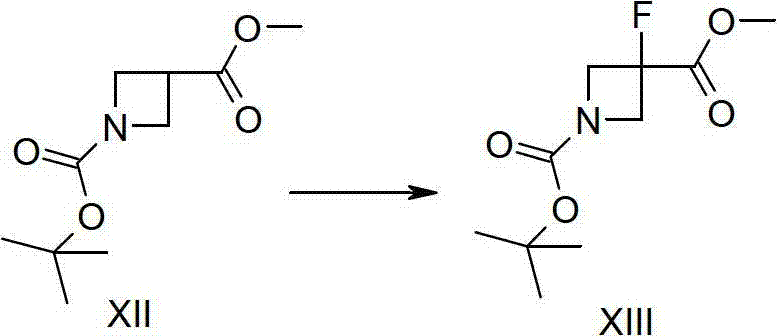
The cornerstone of this synthetic advancement lies in the precise execution of the fluorination step, which transforms the protected azetidine intermediate into the fluorinated derivative. The mechanism involves the generation of a reactive enolate or carbanion species at the 3-position of the azetidine ring. By utilizing strong, non-nucleophilic bases such as lithium hexamethyldisilazide (LiHMDS) or sodium hexamethyldisilazide (NaHMDS) at cryogenic temperatures ranging from -70°C to -90°C, the protocol ensures selective deprotonation without compromising the integrity of the strained four-membered ring. Subsequent treatment with N-fluorobenzenesulfonimide (NFSI) serves as the electrophilic fluorine source, effectively transferring the fluorine atom to the activated carbon center. This specific choice of reagents prevents side reactions such as ring opening or polymerization, which are common pitfalls in azetidine chemistry.
Furthermore, the control of impurities is inherently built into the reaction design through the use of mild hydrolysis conditions in the final step. Unlike oxidative cleavage methods that can generate a myriad of byproducts, the alkaline hydrolysis of the methyl ester is highly chemoselective. It targets only the ester functionality while leaving the sensitive C-F bond and the Boc protecting group intact until specifically required. This selectivity minimizes the formation of structural analogs and degradation products, thereby simplifying the purification process. For quality control teams, this means a cleaner crude profile and a higher likelihood of passing strict impurity specifications without the need for repetitive recrystallization or chromatography, ensuring consistent batch-to-batch quality essential for GMP manufacturing environments.
How to Synthesize 1-Carboxylic Acid Tert-Butyl Ester-3-Fluoro-Azetidine Efficiently
Implementing this synthesis requires careful attention to temperature control and reagent stoichiometry to maximize the benefits of the patented route. The process begins with the protection of the starting amine, followed by the critical low-temperature fluorination, and concludes with hydrolysis. Each stage has been optimized to balance reaction rate with selectivity, ensuring that the final product meets the high standards required for pharmaceutical applications. The following guide outlines the standardized operational parameters derived from the patent embodiments to assist process chemists in replicating this high-efficiency pathway.
- Protect the amine group of methyl azetidine-3-carboxylate hydrochloride using di-tert-butyl dicarbonate under alkaline conditions to form the Boc-protected intermediate.
- Perform electrophilic fluorination at the 3-position of the azetidine ring using N-fluorobenzenesulfonimide (NFSI) and a strong base like LiHMDS at low temperatures.
- Hydrolyze the methyl ester group under alkaline conditions to obtain the final carboxylic acid derivative with high purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers profound strategic advantages beyond simple yield improvements. By collapsing an eight-step sequence into three steps, the manufacturing timeline is drastically compressed, allowing for faster response to market demands and reduced inventory holding costs. The elimination of precious metal catalysts like palladium and ruthenium removes a significant variable cost driver and mitigates the supply risk associated with these geopolitically sensitive materials. Additionally, the simplified workflow reduces the consumption of solvents and energy, aligning with corporate sustainability goals and reducing the environmental footprint of production facilities. These factors collectively contribute to a more resilient and cost-effective supply chain for critical pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The most immediate financial benefit stems from the removal of expensive transition metal catalysts and oxidants from the process bill of materials. Traditional routes relying on palladium and ruthenium incur high raw material costs and necessitate expensive metal recovery or disposal procedures. By switching to an organic fluorination strategy using NFSI, manufacturers can achieve substantial cost savings per kilogram of product. Furthermore, the dramatic increase in overall yield from roughly 2.5% to over 70% means that significantly less starting material is required to produce the same amount of final product, effectively multiplying the purchasing power of the raw material budget and lowering the unit cost of goods sold.
- Enhanced Supply Chain Reliability: Supply chain stability is often compromised by complex syntheses that have multiple potential failure points. An eight-step process offers eight opportunities for yield loss, equipment downtime, or quality deviations. By reducing the synthesis to three robust steps, the probability of successful batch completion increases exponentially. The reagents used in this new method, such as di-tert-butyl dicarbonate and NFSI, are commodity chemicals with stable global supply chains, unlike specialized catalysts that may face shortages. This reliability ensures consistent delivery schedules for downstream API producers, minimizing the risk of production stoppages due to intermediate shortages and fostering stronger long-term partnerships between suppliers and pharmaceutical clients.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to commercial production often reveals hidden bottlenecks, particularly regarding heat transfer and safety. The mild reaction conditions of this new method, operating at ambient or moderately low temperatures without high-pressure hydrogenation, make it inherently safer and easier to scale. The absence of heavy metals simplifies wastewater treatment and reduces the regulatory burden associated with discharging metal-contaminated effluents. This environmental compliance is increasingly critical as global regulations tighten, ensuring that manufacturing facilities can operate continuously without facing shutdowns due to environmental violations, thus securing long-term production capacity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the practical benefits and operational requirements of the new method. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this route for their specific manufacturing needs.
Q: What is the total yield advantage of this new method compared to conventional routes?
A: The novel method achieves a total yield of approximately 74% for the key intermediate and up to 99% for the final conversion, significantly outperforming the conventional 8-step route which yields only 2.46%.
Q: Does this process require expensive transition metal catalysts?
A: No, unlike previous methods that utilized Palladium on Carbon (Pd/C) and Ruthenium trichloride, this optimized route relies on organic reagents like NFSI and standard bases, eliminating heavy metal contamination risks.
Q: Is this synthesis suitable for large-scale commercial production?
A: Yes, the process utilizes mild reaction conditions, common solvents like ethyl acetate and THF, and avoids hazardous oxidants, making it highly scalable and safer for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-Carboxylic Acid Tert-Butyl Ester-3-Fluoro-Azetidine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent theory to commercial reality requires deep technical expertise and robust infrastructure. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the efficiencies promised by CN102731362A are fully realized in practice. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every batch of 1-carboxylic acid tert-butyl ester-3-fluoro-azetidine meets the exacting standards of the global pharmaceutical industry. Our commitment to quality and consistency makes us the preferred partner for companies seeking to optimize their API supply chains.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis can drive value for your organization. By requesting a Customized Cost-Saving Analysis, you can quantify the potential economic benefits of switching to this superior manufacturing route. We encourage you to contact us to obtain specific COA data and route feasibility assessments tailored to your project requirements. Let us collaborate to enhance your supply chain resilience and accelerate your drug development timelines with our high-performance intermediates.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →