Advanced Synthesis of 7-Fluoroimidazo[1,2-A]pyridine for Scalable Antiviral Production
Introduction to Next-Generation Antiviral Intermediate Synthesis
The pharmaceutical industry is constantly seeking more efficient and cost-effective pathways for producing critical heterocyclic scaffolds, particularly those serving as core structures for antiviral therapeutics. A significant breakthrough in this domain is detailed in patent CN112409354B, which discloses a robust synthesis process for 7-fluoroimidazo[1,2-A]pyridine and its key intermediates. This compound class is indispensable in modern medicinal chemistry, yet traditional manufacturing routes have long been plagued by prohibitive costs and harsh reaction conditions. The disclosed technology introduces a strategic pivot by utilizing a quaternary ammonium salt intermediate, effectively bypassing the limitations of previous methodologies. By shifting the synthetic logic to activate the pyridine ring through quaternization prior to fluorination, the process achieves high reactivity under remarkably mild thermal conditions. This innovation not only streamlines the operational workflow but also aligns perfectly with the stringent purity and safety standards required by global regulatory bodies for active pharmaceutical ingredient (API) production.
For R&D directors and process chemists, the implications of this patent are profound, offering a tangible solution to the bottleneck of introducing fluorine atoms into electron-rich heterocyclic systems without resorting to extreme temperatures or precious metal catalysis. The methodology leverages readily available starting materials, specifically 4-chloro-2-aminopyridine, transforming them through a sequence of cyclization, quaternization, fluorination, and deprotection. This approach ensures that the supply chain remains resilient against raw material volatility while maintaining a high degree of chemical fidelity. As we delve deeper into the technical specifics, it becomes evident that this process represents a paradigm shift towards greener, more economical, and scalable chemical manufacturing, positioning it as a preferred choice for reliable pharmaceutical intermediate supplier partnerships aiming to optimize their production pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 7-fluoroimidazo[1,2-A]pyridine has been constrained by two primary technological barriers that hindered large-scale adoption. The first conventional method, as referenced in patent application WO201100790761, relies heavily on the use of palladium catalysts to facilitate the coupling reactions necessary for constructing the fluorinated scaffold. While effective on a small laboratory scale, the reliance on palladium introduces significant economic and logistical burdens, including the high cost of the catalyst itself and the complex downstream processing required to remove trace heavy metals to meet pharmaceutical specifications. Furthermore, the second prevailing method, documented in CN108440402, attempts to circumvent metal catalysis by employing sodium fluoride for direct nucleophilic substitution. However, this approach demands excessively harsh reaction conditions, specifically temperatures reaching up to 180°C, which poses severe challenges for equipment integrity, energy consumption, and safety management in a commercial plant setting.
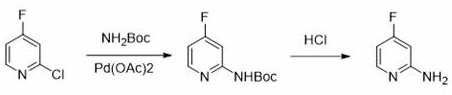
These legacy techniques create a dichotomy for procurement managers and supply chain heads: choose between expensive metal-based routes that complicate purification or endure energy-intensive processes that limit throughput and increase operational risk. The high thermal load required for direct fluorination often leads to increased formation of degradation by-products, complicating the impurity profile and necessitating rigorous and costly purification steps such as silica gel chromatography, which is notoriously difficult to scale. Consequently, the industry has faced a persistent need for a synthesis strategy that balances reactivity with operational simplicity, avoiding both the financial drain of precious metals and the engineering challenges of high-temperature reactors.
The Novel Approach
The innovative process outlined in CN112409354B elegantly resolves these conflicts by introducing a quaternary ammonium salt intermediate as a pivotal activation step. Instead of attempting direct fluorination on the neutral heterocycle, the novel route first converts 7-chloroimidazo[1,2-A]pyridine into a positively charged quaternary salt using halides such as benzyl bromide or p-methoxybenzyl chloride. This structural modification dramatically alters the electronic landscape of the molecule, rendering the carbon-chlorine bond highly susceptible to nucleophilic attack by fluoride ions. As a result, the subsequent fluorination step can be executed at significantly lower temperatures, typically between 90°C and 100°C, which is a substantial reduction from the 180°C required by older methods. This mild condition not only preserves the integrity of the sensitive imidazo-pyridine core but also drastically reduces energy consumption and safety hazards associated with high-pressure, high-temperature operations.
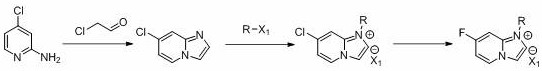
Moreover, this approach eliminates the need for transition metal catalysts entirely, relying instead on abundant and inexpensive inorganic fluoride salts like cesium fluoride or potassium fluoride. The process flow is streamlined into a logical sequence: cyclization of 2-amino-4-chloropyridine with 2-chloroacetaldehyde, followed by quaternization, fluorination, and finally acidic deprotection. Crucially, the intermediate quaternary salt can often be telescoped into the next step without isolation, further reducing solvent usage and processing time. For a reliable pharmaceutical intermediate supplier, this translates to a robust, scalable protocol that delivers high-purity products with a simplified impurity profile, making it ideally suited for cost reduction in API manufacturing where efficiency and compliance are paramount.
Mechanistic Insights into Quaternary Salt Activation and Fluorination
At the heart of this technological advancement lies a sophisticated understanding of physical organic chemistry, specifically the modulation of nucleophilic aromatic substitution (SnAr) reactivity through electronic activation. In the neutral 7-chloroimidazo[1,2-A]pyridine molecule, the electron density distributed across the fused ring system renders the C-Cl bond relatively inert to displacement by fluoride, a poor nucleophile in many contexts. By reacting the nitrogen atom of the imidazole ring with an alkyl or benzyl halide, the molecule is converted into a cationic quaternary ammonium species. This positive charge exerts a powerful electron-withdrawing inductive effect throughout the conjugated system, significantly decreasing the electron density at the C-7 position. This activation lowers the energy barrier for the formation of the Meisenheimer complex during the nucleophilic attack, allowing the fluoride ion to displace the chloride leaving group with high efficiency even at moderate temperatures.
From an impurity control perspective, this mechanism offers distinct advantages over radical-based or metal-catalyzed pathways. The absence of palladium or other transition metals removes the risk of metal-catalyzed side reactions, such as homocoupling or hydrogenolysis, which can generate difficult-to-remove impurities. Furthermore, the specificity of the quaternization step ensures that the fluorination occurs regioselectively at the activated C-7 position, minimizing the formation of isomeric by-products. The use of mild bases and controlled thermal profiles during the initial cyclization step (75-80°C) further ensures that the delicate amino-pyridine starting material is converted cleanly without polymerization or decomposition. This precise control over reaction kinetics and thermodynamics results in a final product with a superior purity profile, reducing the burden on downstream purification units and ensuring consistent quality for high-purity pharmaceutical intermediate batches.
How to Synthesize 7-Fluoroimidazo[1,2-A]pyridine Efficiently
Implementing this synthesis route requires careful attention to stoichiometry and reaction parameters to maximize yield and minimize waste. The process begins with the cyclization of 2-amino-4-chloropyridine and 2-chloroacetaldehyde in the presence of a base such as sodium bicarbonate or triethylamine, typically in an alcoholic or amide solvent system. Following the formation of the chloro-intermediate, the critical quaternization step is performed using a benzyl halide derivative in a polar aprotic solvent like DMF or NMP at elevated temperatures (80-90°C). The resulting quaternary salt is then subjected to fluorination using a fluoride source like cesium fluoride at 90-100°C. Finally, the protecting group is cleaved under acidic conditions to reveal the target molecule. The detailed standardized synthetic steps see the guide below.
- Cyclize 2-amino-4-chloropyridine with 2-chloroacetaldehyde under alkaline conditions at 75-80°C to form 7-chloroimidazo[1,2-A]pyridine.
- React the chloro-intermediate with a halide (e.g., p-methoxybenzyl chloride) to generate the reactive quaternary ammonium salt.
- Perform nucleophilic fluorination using cesium fluoride or similar agents at 90-100°C to substitute the chlorine atom.
- Remove the protecting group under acidic conditions at 60-80°C to yield the final 7-fluoroimidazo[1,2-A]pyridine product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this synthesis process offers transformative benefits that extend far beyond simple chemical yield improvements. The primary advantage lies in the drastic simplification of the supply chain for raw materials. By eliminating the dependency on palladium catalysts, which are subject to significant price volatility and geopolitical supply risks, manufacturers can secure a more stable and predictable cost structure. Additionally, the substitution of high-temperature reactors with standard glass-lined or stainless steel vessels capable of operating at 100°C reduces capital expenditure requirements and maintenance costs. The ability to telescope steps, particularly the conversion of the chloro-intermediate to the quaternary salt and subsequently to the fluoro-species without intermediate isolation, significantly reduces solvent consumption and waste generation, aligning with modern sustainability goals and reducing disposal costs.
- Cost Reduction in Manufacturing: The elimination of expensive noble metal catalysts removes a major cost driver from the bill of materials, while the mild reaction conditions lower energy consumption substantially. Furthermore, the avoidance of silica gel chromatography for purification, which is often required in less selective routes, allows for the use of simpler crystallization or extraction techniques that are far more economical at scale. This cumulative effect results in a significantly reduced cost of goods sold (COGS), enabling competitive pricing strategies for the final API without compromising margin.
- Enhanced Supply Chain Reliability: The starting materials, such as 4-chloro-2-aminopyridine and 2-chloroacetaldehyde, are commodity chemicals with robust global supply networks, ensuring consistent availability and reducing the risk of production stoppages due to raw material shortages. The process tolerance for various bases and solvents provides flexibility in sourcing, allowing procurement teams to negotiate better terms with multiple vendors. This resilience is critical for maintaining continuous production schedules and meeting the just-in-time delivery expectations of downstream pharmaceutical clients.
- Scalability and Environmental Compliance: The mild thermal profile and absence of heavy metals simplify the scale-up process from pilot plant to commercial production, reducing the technical risk associated with technology transfer. The reduced generation of hazardous waste and the potential for solvent recycling contribute to a smaller environmental footprint, facilitating easier regulatory approval and compliance with increasingly stringent environmental, social, and governance (ESG) mandates. This makes the process not only commercially viable but also socially responsible, enhancing the brand reputation of the manufacturer.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and technical disclosures within the patent literature, providing a clear understanding of the process capabilities and limitations. Understanding these details is essential for R&D teams evaluating the feasibility of adopting this route for their specific project timelines and quality requirements.
Q: Why is the quaternary ammonium salt intermediate crucial for this synthesis?
A: The formation of the quaternary ammonium salt significantly increases the electron deficiency of the pyridine ring, thereby activating the C-Cl bond for nucleophilic substitution. This allows fluorination to proceed under much milder conditions (90-100°C) compared to traditional methods requiring temperatures as high as 180°C.
Q: Does this process require expensive transition metal catalysts?
A: No, unlike prior art methods such as those described in WO201100790761 which utilize palladium catalysts, this novel route relies on standard organic bases and fluoride salts. This eliminates the need for costly heavy metal removal steps and reduces the overall production cost significantly.
Q: What are the yield expectations for the final deprotection step?
A: According to the experimental data in patent CN112409354B, the final deprotection step using acids like methanesulfonic acid or trifluoroacetic acid can achieve yields ranging from 77% to 81%, demonstrating high efficiency and robustness for industrial application.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 7-Fluoroimidazo[1,2-A]pyridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient synthesis pathways play in the successful development of antiviral therapeutics. Our team of expert process chemists has thoroughly analyzed the technology disclosed in CN112409354B and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this innovative route to life. We are committed to delivering high-purity intermediates that meet stringent purity specifications, utilizing our rigorous QC labs to ensure every batch conforms to the highest international standards. Our state-of-the-art facilities are equipped to handle the specific solvent systems and thermal requirements of this quaternary salt-mediated fluorination, ensuring a seamless transition from laboratory concept to industrial reality.
We invite pharmaceutical partners to collaborate with us to leverage this cost-effective and scalable technology for their pipeline projects. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise in advanced heterocyclic synthesis can accelerate your drug development timeline and optimize your supply chain economics.