Advanced One-Pot Synthesis of Cilazapril Intermediates for Commercial Scale-Up
The global demand for antihypertensive medications continues to drive the need for efficient, scalable, and cost-effective synthesis routes for key Active Pharmaceutical Ingredients (APIs) and their precursors. Among these, Cilazapril stands out as a potent, long-acting angiotensin-converting enzyme inhibitor (ACEI) widely used in cardiovascular therapy. However, the manufacturing of Cilazapril has historically been plagued by complex synthetic challenges, particularly regarding the stability and stereochemical control of its core heterocyclic intermediates. Patent CN101723902B introduces a transformative methodology that addresses these critical bottlenecks by utilizing a direct hydrogenation and in-situ salt formation strategy. This technical breakthrough not only streamlines the production workflow but also significantly enhances the stability of the final intermediate, making it a highly attractive candidate for commercial scale-up. For pharmaceutical manufacturers seeking a reliable cilazapril intermediate supplier, understanding the nuances of this patented process is essential for optimizing supply chains and reducing overall production costs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic pathways for Cilazapril intermediates often involve the isolation of the free base form, specifically (1S)-hexahydro-pyridazine-3-tert-butyl carboxylic ester. As detailed in the background art of the patent, this free base compound exhibits extreme instability when exposed to atmospheric conditions. During standard filtration and purification procedures, a significant portion of the material undergoes unwanted oxidation, converting into azo-compounds and various other by-products. This degradation not only drastically reduces the overall yield of the finished API but also complicates the purification process, requiring rigorous and expensive chromatographic steps to remove impurities. Furthermore, the multi-step nature of conventional routes, which often necessitates separate protection and deprotection stages followed by isolated intermediate handling, increases the operational complexity and the risk of stereochemical erosion. These factors collectively contribute to higher manufacturing costs and longer lead times, creating substantial friction in the supply chain for high-purity pharmaceutical intermediates.
The Novel Approach
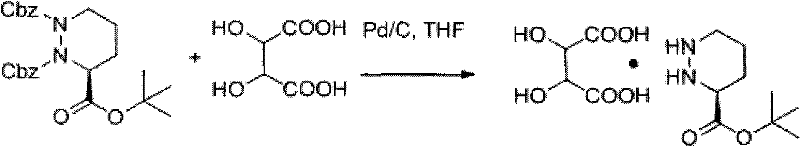
In stark contrast to the precarious handling of the free base, the novel approach disclosed in the patent employs a clever one-pot strategy that bypasses the isolation of the unstable species entirely. By introducing L-tartaric acid directly into the hydrogenation reactor alongside the protected precursor, the process facilitates the simultaneous removal of benzyl protecting groups and the immediate formation of the stable L-tartrate salt. This in-situ trapping mechanism effectively shields the reactive hydrazine moiety from oxidative degradation, ensuring that the intermediate remains in its stable salt form throughout the workup. The result is a white solid that can be easily purified through simple crystallization from n-butanol, eliminating the need for complex separation techniques. This methodological shift represents a paradigm change in cost reduction in pharmaceutical intermediates manufacturing, as it consolidates multiple unit operations into a single, robust reaction vessel, thereby minimizing solvent usage, labor hours, and potential yield losses associated with intermediate transfers.
Mechanistic Insights into Pd/C-Catalyzed Hydrogenolysis and Salt Formation
The core of this innovative synthesis lies in the precise orchestration of catalytic hydrogenolysis and acid-base chemistry within a single reaction medium. The process utilizes Palladium on Carbon (Pd/C) as a heterogeneous catalyst to cleave the benzyl carbamate (Cbz) protecting groups from the nitrogen atoms of the hexahydro-pyridazine ring. Under controlled conditions of 65°C to 75°C and a hydrogen pressure of 4 to 5 atm, the catalyst facilitates the addition of hydrogen across the benzyl-oxygen bonds, releasing toluene and carbon dioxide while freeing the amine functionalities. Crucially, the presence of L-tartaric acid in the reaction mixture ensures that as soon as the basic nitrogen centers are liberated, they are immediately protonated and paired with the tartrate anion. This rapid salt formation lowers the energy of the system and prevents the free amine from participating in side reactions, such as the aforementioned oxidation to azo-species. The choice of solvent, typically tetrahydrofuran (THF), ethanol, or methanol, plays a vital role in solubilizing both the organic precursor and the acidic salt former, creating a homogeneous environment that maximizes reaction kinetics and ensures uniform crystal growth during the subsequent cooling phase.
From an impurity control perspective, this mechanism offers distinct advantages over stepwise approaches. In traditional methods, the exposure of the free base to air during filtration creates a window of vulnerability where oxidative impurities can form and propagate. By maintaining the intermediate in the salt form from the moment of generation, the novel route effectively closes this window. The resulting L-tartrate salt possesses a defined crystal lattice structure that inherently rejects impurities during crystallization, leading to a product with superior chemical and optical purity. This is particularly critical for chiral drugs like Cilazapril, where the presence of the wrong enantiomer or diastereomer can have significant pharmacological consequences. The ability to consistently produce the (S)-configuration with high fidelity underscores the robustness of this catalytic system, providing R&D directors with the confidence that the process can meet stringent regulatory specifications for commercial API production without the need for extensive downstream remediation.
How to Synthesize (S)-Hexahydro-pyridazine-3-tert-butyl Carboxylic Ester L-Tartrate Efficiently
Implementing this synthesis requires careful attention to reaction parameters to maximize the benefits of the one-pot design. The process begins by charging a hydrogenation reactor with the protected precursor, (1S)-1,2-di-benzoyloxycarbonyl-3-tert-butyl hexahydro pyridazine-1,2,3-tricarboxylic ester, and L-tartaric acid in a molar ratio preferably around 1:1.2 to ensure complete salt formation. A suitable solvent such as THF is added along with the Pd/C catalyst, and the system is pressurized with hydrogen. The reaction is exothermic and requires precise temperature control to maintain the optimal range of 65°C to 75°C, ensuring complete deprotection without degrading the sensitive heterocyclic core. Once hydrogen uptake ceases, indicating the completion of the reaction, the mixture is cooled to induce crystallization. The detailed standardized synthesis steps, including specific washing protocols and crystallization rates, are outlined below to guide process engineers in replicating this high-yield pathway.
- Charge a hydrogenation reactor with the protected precursor (1S)-1,2-di-benzoyloxycarbonyl-3-tert-butyl hexahydro pyridazine-1,2,3-tricarboxylic ester, L-tartaric acid, a solvent such as THF or ethanol, and a Pd/C catalyst.
- Control the reaction temperature between 65°C and 75°C and maintain hydrogen pressure at 4 to 5 atm until hydrogen uptake ceases, ensuring complete deprotection.
- Cool the mixture to 0°C, filter to remove the catalyst, wash the cake, concentrate the filtrate, and crystallize the resulting white solid from n-butanol to obtain the pure L-tartrate salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology translates into tangible strategic advantages that extend beyond mere technical feasibility. The consolidation of reaction steps into a single vessel significantly reduces the capital expenditure required for processing equipment, as fewer reactors and isolation units are needed to achieve the same output. Furthermore, the elimination of unstable intermediate handling reduces the risk of batch failures and safety incidents, leading to a more predictable and reliable production schedule. This stability is crucial for maintaining continuous supply lines to downstream API manufacturers, who depend on consistent quality and timely delivery to meet their own production targets. By mitigating the risks associated with oxidative degradation and complex purifications, this route offers a pathway to substantial cost savings and enhanced operational efficiency.
- Cost Reduction in Manufacturing: The primary driver for cost optimization in this process is the drastic simplification of the workflow. By avoiding the isolation of the unstable free base, manufacturers eliminate the need for specialized inert atmosphere handling equipment and the associated operational overheads. The high yields reported in the patent embodiments, ranging from roughly 76% to 81%, indicate a highly atom-economical process that minimizes raw material waste. Additionally, the use of common, recyclable solvents like THF and n-butanol, combined with a reusable heterogeneous catalyst, further drives down the variable costs per kilogram. This efficiency allows suppliers to offer competitive pricing structures without compromising on margin, making it an economically viable solution for large-scale generic drug production.
- Enhanced Supply Chain Reliability: Supply chain resilience is heavily dependent on the robustness of the underlying chemistry. The sensitivity of the conventional free base intermediate to air and moisture creates a fragile link in the supply chain, where minor deviations in handling can lead to significant quality deviations or total batch loss. The new tartrate salt form, being dimensionally stable and easy to preserve, removes this fragility. It allows for safer storage and transportation, reducing the likelihood of spoilage during logistics. This stability ensures that inventory can be held with confidence, enabling suppliers to buffer against demand fluctuations and guaranteeing shorter lead times for high-purity pharmaceutical intermediates to their global clientele.
- Scalability and Environmental Compliance: Scaling chemical processes often amplifies environmental and safety challenges, but this route is inherently designed for scalability. The hydrogenation conditions (4-5 atm) are moderate and easily manageable in standard industrial hydrogenators, posing minimal safety risks compared to high-pressure alternatives. Moreover, the reduction in solvent swaps and purification steps leads to a lower overall E-factor (environmental factor), meaning less waste is generated per unit of product. The ability to filter and wash the catalyst cake efficiently also facilitates the recovery and recycling of precious metals, aligning with modern green chemistry principles and helping manufacturers meet increasingly stringent environmental regulations without costly retrofits.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. They are derived from the specific technical disclosures and beneficial effects highlighted in the patent documentation, aiming to clarify the operational benefits for potential partners. Understanding these details is key to evaluating the feasibility of integrating this intermediate into your existing supply chain.
Q: Why is the L-tartrate salt form preferred over the free base for this intermediate?
A: The free base form, (1S)-hexahydro-pyridazine-3-tert-butyl carboxylic ester, is extremely unstable in air and prone to oxidation into azo-compounds during filtration. Converting it directly to the L-tartrate salt stabilizes the molecule, preventing degradation and simplifying purification.
Q: What catalysts are suitable for this hydrogenation process?
A: While Palladium on Carbon (Pd/C) is the preferred catalyst for efficient hydrogenolysis in the patented method, the process is robust enough to accommodate other catalysts such as iron, zinc, or nickel, offering flexibility in procurement.
Q: What yields can be expected from this synthetic route?
A: The patent documentation reports consistent yields ranging from approximately 76% to 81% across multiple embodiments, demonstrating a highly efficient and reproducible process suitable for industrial application.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cilazapril Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory patent to commercial reality requires more than just chemical knowledge; it demands engineering excellence and rigorous quality assurance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising yields and purity profiles demonstrated in patent CN101723902B can be reliably reproduced on an industrial scale. We are equipped with state-of-the-art hydrogenation facilities and stringent purity specifications enforced by our rigorous QC labs, guaranteeing that every batch of cilazapril intermediate meets the exacting standards required for global regulatory filings. Our commitment to technical precision ensures that the stability and cost advantages of this one-pot route are fully realized for our partners.
We invite pharmaceutical companies and contract manufacturers to explore how this advanced synthesis route can optimize their Cilazapril supply chain. By leveraging our expertise in process development, we can provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and logistical constraints. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our capabilities align with your strategic sourcing goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →