Advanced One-Pot Synthesis of Tetrahydroquinoline Chroman Polycyclic Compounds for Oncology Drug Development
The pharmaceutical industry is constantly seeking robust and efficient methodologies to construct complex nitrogen-containing heterocycles, which serve as the backbone for numerous bioactive molecules. Patent CN115232140B introduces a groundbreaking approach to synthesizing tetrahydroquinoline and chroman polycyclic compounds, a class of structures with profound implications for oncology drug development. This technology leverages a novel one-pot cascade reaction strategy that effectively merges substituted quinoline salts, ethyl 2,3-butadienoate, and 8-hydroxyisoquinoline. By bypassing the traditional multi-step sequences often required for such intricate architectures, this method addresses critical bottlenecks in medicinal chemistry pipelines. The resulting polycyclic scaffolds are not only structurally diverse but also exhibit promising antitumor properties, making them highly valuable candidates for further pharmacological evaluation.
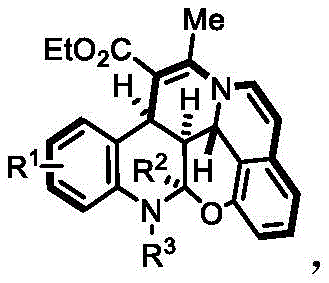
For R&D directors focused on pipeline acceleration, the ability to access these complex cores rapidly is a significant strategic advantage. The patent details a versatile synthetic route where R1 can be hydrogen, a halogen, or an alkyl group, while R2 and R3 offer further diversification opportunities. This structural flexibility allows for the rapid generation of analog libraries essential for structure-activity relationship (SAR) studies. Furthermore, the methodology operates under relatively mild conditions, utilizing common organic solvents and bases, which simplifies the technical requirements for implementation in standard laboratory settings. The integration of the chroman skeleton alongside the tetrahydroquinoline core creates a rigid, three-dimensional framework that is increasingly prized in modern drug design for its ability to improve binding affinity and metabolic stability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of multi-substituted tetrahydroquinoline polycyclic compounds has been fraught with challenges, particularly regarding selectivity and operational complexity. Conventional strategies often rely on the dearomatization of quinoline salts, a process that is theoretically direct but practically difficult due to the presence of multiple reactive sites on the quinoline ring. This lack of differentiation frequently leads to poor regioselectivity and stereoselectivity, resulting in complex mixtures of isomers that are difficult and costly to separate. Moreover, traditional routes may require harsh reaction conditions, expensive transition metal catalysts, or protection-deprotection sequences that increase the step count and reduce overall atom economy. These inefficiencies translate directly into higher production costs and longer lead times, creating substantial barriers for the commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology described in CN115232140B utilizes a highly efficient tandem cyclization strategy that constructs the polycyclic framework in a single operational step. By reacting various substituted quinoline salts with ethyl 2,3-butadienoate and 8-hydroxyisoquinoline in the presence of a base, the system achieves high bond-forming and ring-forming efficiency with excellent selectivity. The reaction proceeds smoothly in organic solvents such as chloroform at temperatures ranging from 25°C to 100°C, with a preferred embodiment at 60°C. This mild thermal profile minimizes energy consumption and reduces the risk of thermal degradation of sensitive functional groups. Crucially, the intermediate products do not require isolation or purification, allowing the reaction to proceed directly to the final target, which is easily separated via standard column chromatography.
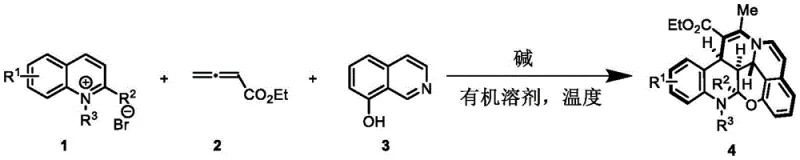
The visual representation of this transformation underscores the elegance of the design, where three distinct components converge to form a complex fused ring system with multiple stereocenters controlled effectively. This one-pot nature drastically simplifies the workflow, reducing the manpower and solvent usage associated with intermediate workups. For procurement and supply chain teams, this translates to a more streamlined manufacturing process that is less prone to yield losses between steps. The broad substrate scope demonstrated in the patent, accommodating various halogens and alkyl groups, ensures that this platform technology can be adapted to synthesize a wide array of derivatives without needing to reinvent the wheel for each new analog, thereby enhancing the overall agility of the drug discovery process.
Mechanistic Insights into Base-Catalyzed Tandem Cyclization
The success of this synthesis lies in the precise orchestration of nucleophilic attacks and cyclization events facilitated by the base catalyst. The reaction initiates with the activation of the nucleophilic species, likely involving the deprotonation of the 8-hydroxyisoquinoline or the activation of the allenoate ester, which then attacks the electrophilic quinoline salt. This initial addition triggers a cascade of intramolecular cyclizations that construct the tetrahydroquinoline and chroman rings simultaneously. The use of bases such as N-methylmorpholine, triethylamine, or inorganic carbonates provides the necessary alkalinity to drive the reaction forward without promoting unwanted side reactions. The mechanistic pathway is designed to favor the formation of the thermodynamically stable polycyclic product, ensuring high stereocontrol throughout the process. This level of mechanistic understanding allows chemists to fine-tune reaction parameters, such as base strength and solvent polarity, to optimize outcomes for specific substrate combinations.
From an impurity control perspective, the high selectivity of this cascade reaction is a major benefit. Because the reaction pathway is well-defined and the intermediates are transient, the formation of byproducts is minimized compared to stepwise approaches where isolation errors can accumulate. The patent data shows that even with diverse substituents like fluorine, bromine, or nitro groups, the reaction maintains good fidelity, producing the desired scaffolds with consistent quality. This robustness is critical for GMP manufacturing, where impurity profiles must be tightly controlled to meet regulatory standards. The ability to achieve high purity directly from the reaction mixture, followed by a simple chromatographic purification, reduces the burden on downstream processing units. Consequently, this method supports the production of high-purity pharmaceutical intermediates that are ready for subsequent biological testing or further functionalization with minimal additional processing.
How to Synthesize Tetrahydroquinoline Chroman Derivatives Efficiently
Implementing this synthesis protocol requires careful attention to reagent stoichiometry and reaction monitoring to ensure optimal conversion. The standard procedure involves mixing the substituted quinoline salt, ethyl 2,3-butadienoate, and 8-hydroxyisoquinoline in a suitable organic solvent, with chloroform being the preferred medium for its solubility properties and reaction performance. A base, such as N-methylmorpholine, is added to initiate the cascade, and the mixture is heated to the target temperature, typically around 60°C. Reaction progress is monitored using thin-layer chromatography (TLC) until the starting materials are fully consumed, which usually occurs within 0.5 to 2 hours depending on the specific substrate electronics. Once complete, the crude mixture is subjected to column chromatography using a petroleum ether and ethyl acetate gradient to isolate the pure polycyclic product. Detailed standardized synthesis steps are provided in the guide below to ensure reproducibility across different batches and scales.
- Combine substituted quinoline salts, ethyl 2,3-butadienoate, and 8-hydroxyisoquinoline in an organic solvent such as chloroform.
- Add a base catalyst, preferably N-methylmorpholine, to the reaction mixture to initiate the cascade cyclization.
- Stir the mixture at a temperature between 25°C and 100°C for 0.5 to 2 hours, then purify the target product via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits in terms of cost efficiency and supply reliability. The primary drivers of cost reduction in pharmaceutical intermediates manufacturing are often the number of synthetic steps and the cost of raw materials. This one-pot method significantly reduces the step count by eliminating intermediate isolation, which in turn lowers solvent consumption, waste generation, and labor costs. Furthermore, the starting materials—substituted quinoline salts, ethyl 2,3-butadienoate, and 8-hydroxyisoquinoline—are commercially available and relatively inexpensive compared to specialized organometallic reagents. The absence of precious metal catalysts removes the need for expensive metal scavenging processes and reduces the risk of heavy metal contamination in the final API, simplifying the regulatory compliance landscape.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts and the reduction of synthetic steps lead to substantial cost savings. By avoiding expensive palladium or rhodium catalysts often used in cross-coupling reactions, the material cost per kilogram of the intermediate is drastically lowered. Additionally, the mild reaction conditions reduce energy expenditures associated with heating and cooling, contributing to a lower overall carbon footprint and operational expense. The simplified workup procedure further decreases the consumption of silica gel and solvents during purification, enhancing the economic viability of large-scale production.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals rather than bespoke reagents ensures a stable and resilient supply chain. Since the key building blocks are widely produced by multiple chemical suppliers, the risk of supply disruption due to single-source dependency is minimized. The robustness of the reaction conditions, which tolerate a range of temperatures and solvents, provides flexibility in manufacturing scheduling and equipment utilization. This adaptability allows for faster response times to fluctuating demand, ensuring that critical oncology intermediates can be delivered consistently without lengthy lead times.
- Scalability and Environmental Compliance: The protocol is inherently scalable, having been demonstrated effectively in batch reactors with straightforward temperature control. The use of common organic solvents like chloroform or ethyl acetate facilitates easy solvent recovery and recycling, aligning with green chemistry principles. The high atom economy of the cascade reaction means less waste is generated per unit of product, simplifying waste treatment protocols and reducing environmental compliance costs. This makes the process attractive for contract development and manufacturing organizations (CDMOs) looking to offer sustainable and scalable solutions for complex drug candidates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these tetrahydroquinoline chroman polycyclic compounds. The answers are derived directly from the experimental data and beneficial effects outlined in the patent documentation, providing clarity on the method's capabilities and limitations. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this technology into their existing drug development workflows. The information covers aspects ranging from reaction optimization to biological potential, ensuring a comprehensive overview for decision-makers.
Q: What are the primary advantages of this synthesis method over traditional dearomatization?
A: This method offers superior regioselectivity and stereoselectivity compared to conventional quinoline salt dearomatization, which often suffers from poor control. Additionally, it utilizes a one-pot cascade strategy that eliminates the need for intermediate isolation, significantly streamlining the workflow.
Q: What are the optimal reaction conditions for maximizing yield?
A: The patent data indicates that using chloroform as the solvent and N-methylmorpholine as the base at 60°C provides excellent results. The reaction typically completes within 0.5 to 2 hours, offering a rapid turnaround time for complex scaffold generation.
Q: Do these compounds demonstrate biological activity relevant to drug discovery?
A: Yes, specific derivatives such as compounds 4a, 4c, and 4j have shown significant cytotoxicity against human lung cancer (A549) and leukemia (K562) cell lines, with IC50 values comparable to the standard drug cisplatin, highlighting their potential as antitumor agents.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tetrahydroquinoline Chroman Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing high-quality, complex intermediates for the development of next-generation therapeutics. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from benchtop discovery to full-scale manufacturing. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of tetrahydroquinoline chroman derivatives meets the highest industry standards. We are committed to supporting your oncology drug development programs with reliable supply and technical excellence.
We invite you to collaborate with us to leverage this advanced synthetic technology for your specific needs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your project's volume and purity requirements. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. By partnering with us, you gain access to a supply chain partner dedicated to accelerating your timeline and optimizing your manufacturing costs for high-purity oncology intermediates.