Advanced Synthesis of Epristeride Impurity for High-Purity Reference Standards and Quality Control
The pharmaceutical industry faces increasing regulatory pressure to fully characterize and control impurities within Active Pharmaceutical Ingredients (APIs), particularly for complex steroid-based therapeutics like Epristeride. Patent CN111362999B, published in March 2022, addresses a critical gap in the market by disclosing a robust preparation method for a specific Epristeride impurity, chemically known as 17 beta- (N-tert-butyl-amino-formyl) androst-4-en-3-one-5-hydroxy. Prior to this innovation, the lack of a standardized synthesis method hindered the ability of quality control laboratories to accurately identify and quantify this degradation product, posing risks to drug safety and efficacy. This technical insight report analyzes the proprietary oxidation strategy detailed in the patent, highlighting its potential to streamline the production of high-purity reference standards essential for global regulatory compliance.
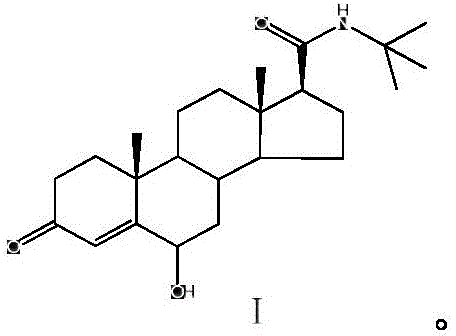
For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediate supplier, understanding the structural integrity of this impurity is paramount. The molecule retains the core androstane skeleton but features specific functionalization at the C-5 position, distinguishing it from the parent drug. The ability to synthesize this exact congener allows for the development of validated HPLC methods that can separate the impurity from the main API peak with high resolution. As we delve deeper into the technical specifics, it becomes evident that this patent offers more than just a chemical recipe; it provides a strategic pathway for enhancing the overall quality assurance framework of Epristeride manufacturing, ensuring that final formulations meet the rigorous safety standards demanded by international health authorities.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of specific steroid impurities has been plagued by significant technical hurdles, often requiring multi-step sequences with harsh reagents that compromise yield and stereochemical integrity. In the specific case of Epristeride, the background art indicates that no viable method existed for synthesizing this particular 5-hydroxy impurity, forcing manufacturers to rely on isolation from degradation studies which is inefficient and yields insufficient quantities for comprehensive analysis. Conventional oxidation methods for steroids often suffer from poor regioselectivity, leading to complex mixtures of over-oxidized byproducts that are difficult to separate. Furthermore, traditional approaches might utilize expensive or hazardous catalysts that introduce heavy metal residues, necessitating additional purification steps that drive up costs and extend lead times. The absence of a dedicated synthetic route meant that impurity profiling was largely reactive rather than proactive, leaving potential quality gaps in the supply chain of this critical benign prostatic hyperplasia medication.
The Novel Approach
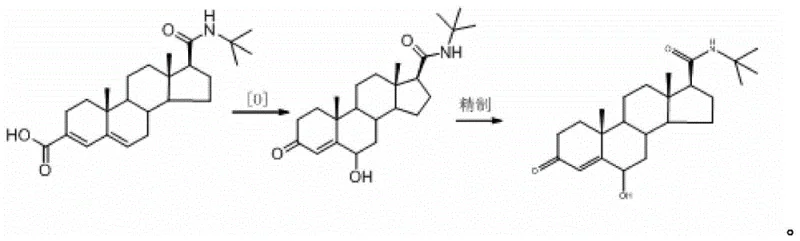
The methodology outlined in patent CN111362999B represents a paradigm shift by utilizing a direct, one-pot oxidative transformation that converts Epristeride directly into the target impurity with remarkable efficiency. By employing common oxidants such as hydrogen peroxide or tert-butyl hydroperoxide in readily available organic solvents like methanol or ethanol, the process eliminates the need for exotic reagents or complex catalytic systems. The reaction proceeds under exceptionally mild conditions, typically between 10°C and 30°C, which minimizes thermal degradation and preserves the sensitive steroid backbone. This approach not only simplifies the operational workflow but also significantly enhances safety profiles by avoiding high-pressure or high-temperature reactors. The subsequent purification via recrystallization ensures that the final product achieves a purity level exceeding 96%, making it immediately suitable for use as a certified reference material without further chromatographic polishing.
Mechanistic Insights into Selective Oxidative Functionalization
The core of this innovation lies in the selective oxidative functionalization of the steroid A-ring, transforming the conjugated diene system of Epristeride into the specific 5-hydroxy-4-en-3-one motif found in the impurity. While the exact mechanistic pathway may involve initial epoxidation or allylic oxidation followed by rearrangement, the net result is the precise installation of the hydroxyl group at the C-5 position while maintaining the ketone at C-3 and the amide side chain at C-17. The use of acidic additives, such as acetic acid or hydrochloric acid, in conjunction with peroxides likely facilitates the activation of the oxidant and directs the regioselectivity towards the desired position. This controlled reactivity is crucial for preventing the formation of isomeric byproducts that could complicate the analytical landscape. Understanding this mechanism allows process chemists to fine-tune reaction parameters, such as stoichiometry and solvent polarity, to maximize the conversion rate while suppressing side reactions.
From an impurity control perspective, the mechanism ensures that the generated reference standard is structurally identical to the degradation product formed during shelf-life storage or stress testing of the API. This identity is confirmed through comprehensive spectroscopic analysis, including 1H-NMR, 13C-NMR, and LC-MS, which match the theoretical values for the proposed structure. The ability to replicate the degradation pathway synthetically means that manufacturers can spike their samples with known quantities of this impurity to validate their detection limits. For technical teams, this mechanistic clarity translates into confidence; they know that the reference standard behaves predictably in analytical columns, providing accurate retention times and response factors. This level of characterization is indispensable for filing Drug Master Files (DMFs) and responding to regulatory queries regarding impurity fate and purge.
How to Synthesize Epristeride Impurity Efficiently
The synthesis protocol described in the patent is designed for scalability and reproducibility, making it an ideal candidate for technology transfer from the lab to pilot plant operations. The process begins with the dissolution of Epristeride in a primary organic solvent, followed by the controlled addition of the oxidant system. Reaction monitoring is straightforward, typically requiring 30 to 50 hours to reach completion, after which the mixture is quenched with a reducing agent like sodium thiosulfate to destroy excess peroxide. The workup involves standard liquid-liquid extraction techniques to isolate the crude product, which is then subjected to a recrystallization step to achieve the final purity specifications. For detailed operational parameters, including specific mass ratios and temperature ramps, please refer to the standardized guide below.
- React Epristeride with an oxidant such as hydrogen peroxide in an organic solvent like methanol at mild temperatures (10-30°C) for 30-50 hours.
- Quench the reaction with sodium thiosulfate, extract with dichloromethane, and concentrate to obtain the crude oily impurity.
- Recrystallize the oily residue using a solvent such as ethyl acetate or acetonitrile to isolate the pure white solid impurity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis route offers tangible benefits that extend beyond mere technical feasibility. The reliance on commodity chemicals such as hydrogen peroxide, methanol, and ethyl acetate drastically reduces the raw material cost base compared to routes requiring specialized catalysts or cryogenic conditions. This simplification of the bill of materials (BOM) mitigates supply risk, as these solvents and reagents are globally available from multiple vendors, ensuring continuity of supply even during market fluctuations. Furthermore, the mild reaction temperatures reduce energy consumption for heating and cooling, contributing to a lower carbon footprint and aligning with corporate sustainability goals. The high yield and purity reported in the examples suggest that waste generation is minimized, lowering the costs associated with effluent treatment and disposal.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of ambient temperature conditions significantly lowers the operational expenditure (OPEX) associated with producing this reference standard. By avoiding complex purification technologies like preparative HPLC and relying instead on crystallization, the process achieves substantial cost savings in both equipment usage and labor. The high atom economy of the direct oxidation step means that less starting material is wasted, further driving down the cost per gram of the final impurity standard. These efficiencies allow suppliers to offer competitive pricing for high-purity reference materials, making quality control more affordable for generic drug manufacturers.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions ensures consistent batch-to-batch quality, which is critical for maintaining a reliable supply of reference standards. Since the process does not depend on sensitive reagents that degrade quickly or require special shipping conditions, inventory management becomes more predictable. The scalability of the method means that suppliers can rapidly ramp up production to meet sudden spikes in demand from regulatory audits or new drug filings. This agility reduces lead times for customers who need urgent access to qualified impurities for method validation, thereby preventing delays in their own regulatory submission timelines.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively from gram to multi-gram scales in the patent examples without loss of efficiency. The use of green oxidants like hydrogen peroxide, which decomposes into water and oxygen, minimizes the generation of hazardous byproducts, simplifying environmental compliance and waste disposal protocols. Solvent recovery systems can be easily integrated to recycle methanol and ethyl acetate, further enhancing the environmental profile of the manufacturing process. This alignment with green chemistry principles not only reduces regulatory burden but also enhances the brand reputation of the manufacturer as a responsible partner in the pharmaceutical supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this Epristeride impurity. These answers are derived directly from the experimental data and claims presented in patent CN111362999B, ensuring accuracy and relevance for industry professionals. Understanding these details helps stakeholders make informed decisions about integrating this reference standard into their quality control workflows.
Q: What is the primary advantage of this new synthesis method for Epristeride impurity?
A: The primary advantage is the simplicity and mildness of the reaction conditions. Unlike previous non-existent or complex methods, this process uses common oxidants like hydrogen peroxide at low temperatures (10-30°C), ensuring high safety and easy control while achieving purity levels exceeding 96%.
Q: Which oxidants are compatible with this synthetic route?
A: The patent specifies a range of effective oxidants including hydrogen peroxide, tert-butyl hydroperoxide, m-chloroperoxybenzoic acid, and peracetic acid. Hydrogen peroxide is particularly preferred for its cost-effectiveness and environmental profile when used with acidic additives.
Q: How does this method support regulatory compliance for Epristeride APIs?
A: By providing a reliable source of the specific impurity (17 beta- (N-tert-butyl-amino-formyl) androst-4-en-3-one-5-hydroxy), manufacturers can use it as a reference substance. This allows for precise quantification and control of impurities in bulk drugs, meeting stringent pharmacopoeia requirements.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Epristeride Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality impurities play in ensuring the safety and efficacy of pharmaceutical products. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency and precision. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify the identity and purity of every batch we produce. Whether you require custom synthesis of complex steroid intermediates or large-scale supply of reference standards, our infrastructure is designed to support your most demanding projects.
We invite you to contact our technical procurement team to discuss how we can support your specific needs for Epristeride impurities and related compounds. By partnering with us, you gain access to a Customized Cost-Saving Analysis that identifies opportunities to optimize your supply chain without compromising on quality. Reach out today to request specific COA data and route feasibility assessments, and let us demonstrate why we are the preferred partner for global pharmaceutical companies seeking reliability and excellence.