Advanced Synthesis of 5-Trifluoromethylpyrimidine Derivatives for Oncology Drug Discovery
Advanced Synthesis of 5-Trifluoromethylpyrimidine Derivatives for Oncology Drug Discovery
The pharmaceutical industry is constantly seeking novel scaffolds to overcome drug resistance in oncology, particularly within the realm of kinase inhibitors. Patent CN111454219A introduces a significant advancement in this field by disclosing a series of 5-trifluoromethylpyrimidine derivatives characterized by a unique substitution pattern that enhances water solubility and tumor cell inhibition. As a leading reliable pharmaceutical intermediate supplier, we recognize that the structural novelty of these compounds lies in the specific arrangement of the trifluoromethyl group at the 5-position coupled with diverse amine substitutions at the 2-position. This configuration is critical for maintaining binding affinity against mutated kinase targets while improving pharmacokinetic profiles. The patent details a robust methodology that not only expands the chemical space available for drug discovery but also offers a practical pathway for the commercial scale-up of complex pharmaceutical intermediates. By leveraging this technology, research teams can accelerate the development of next-generation anti-tumor agents that address the growing challenge of acquired resistance in cancer therapy.
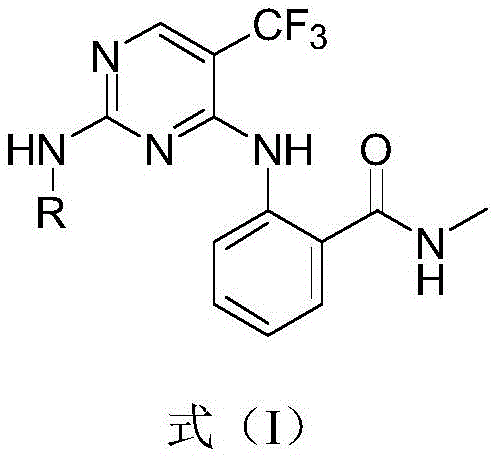
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of polysubstituted pyrimidines often suffers from poor regioselectivity, especially when introducing multiple nitrogen-containing substituents onto the heterocyclic ring. Conventional nucleophilic aromatic substitution (SNAr) reactions on dichloropyrimidines can lead to mixtures of mono- and di-substituted products, requiring tedious purification steps that drastically reduce overall yield. Furthermore, harsh reaction conditions frequently employed in older methodologies can degrade sensitive functional groups or lead to the hydrolysis of the trifluoromethyl-containing scaffold. These inefficiencies create bottlenecks in the supply chain, increasing the cost of goods and extending the lead time for high-purity intermediates needed for preclinical studies. The inability to selectively functionalize the 2-position after establishing the 4-position linkage has historically limited the speed at which medicinal chemists can explore structure-activity relationships (SAR) around the pyrimidine core.
The Novel Approach
The methodology described in the patent overcomes these hurdles through a strategic two-step sequence that maximizes both selectivity and efficiency. The process begins with a mild nucleophilic substitution at the 4-position of 2,4-dichloro-5-trifluoromethylpyrimidine, taking advantage of the electronic activation provided by the ring nitrogens and the trifluoromethyl group. This is followed by a palladium-catalyzed Buchwald-Hartwig amination at the remaining 2-chloro position. This transition metal-catalyzed cross-coupling allows for the introduction of a wide variety of sterically hindered and electronically diverse amines that would be difficult to install via direct SNAr. 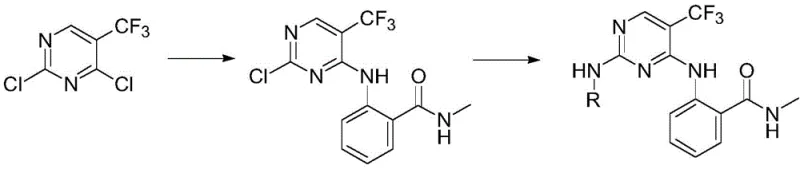 This approach ensures that the core scaffold remains intact while providing unparalleled flexibility in modifying the side chains, thereby facilitating cost reduction in API manufacturing by streamlining the synthetic route and minimizing waste generation associated with isomer separation.
This approach ensures that the core scaffold remains intact while providing unparalleled flexibility in modifying the side chains, thereby facilitating cost reduction in API manufacturing by streamlining the synthetic route and minimizing waste generation associated with isomer separation.
Mechanistic Insights into Pd-Catalyzed Buchwald-Hartwig Amination
The success of this synthetic strategy hinges on the precise control of the catalytic cycle during the second step. The reaction utilizes a palladium source, specifically Pd(OAc)2, in conjunction with the bidentate ligand xantphos. The large bite angle of xantphos is crucial for stabilizing the active palladium species and facilitating the reductive elimination step, which is often the rate-determining step in the formation of C-N bonds on electron-deficient heterocycles. The mechanism involves the oxidative addition of the aryl chloride bond to the Pd(0) species, followed by coordination and deprotonation of the amine substrate by the base, sodium tert-butoxide (NaOtBu). The resulting amido-palladium complex then undergoes reductive elimination to release the desired product and regenerate the catalyst. This catalytic system is highly tolerant of the trifluoromethyl group and the existing amide functionality at the 4-position, demonstrating excellent chemoselectivity.
From an impurity control perspective, the choice of solvent and base plays a pivotal role. The use of anhydrous 1,4-dioxane under an inert argon atmosphere prevents the hydrolysis of the chloro-intermediate, which is a common side reaction in aqueous or protic media. Additionally, the mild basicity of the system compared to stronger alkoxides helps prevent potential degradation of the benzamide moiety. By optimizing the molar ratios of the catalyst, ligand, and base, the process minimizes the formation of dehalogenated byproducts and homocoupling impurities. This rigorous control over the reaction parameters ensures that the final high-purity pharmaceutical intermediates meet the stringent quality standards required for biological testing and subsequent clinical development, reducing the burden on downstream purification processes.
How to Synthesize 5-Trifluoromethylpyrimidine Derivatives Efficiently
The preparation of these potent anti-tumor agents follows a logical progression from readily available starting materials to complex final products. The initial step involves the condensation of 2,4-dichloro-5-trifluoromethylpyrimidine with 2-amino-N-methylbenzamide in ethanol, utilizing sodium bicarbonate as a mild base to scavenge the generated HCl. This reaction proceeds smoothly at room temperature to yield the key chloro-intermediate, which serves as the divergent point for library synthesis. The subsequent coupling step requires careful attention to moisture exclusion and temperature control to ensure high conversion. For detailed operational parameters and safety considerations regarding the handling of palladium catalysts and organic solvents, please refer to the standardized protocol below.
- Perform selective nucleophilic substitution of 2,4-dichloro-5-trifluoromethylpyrimidine with 2-amino-N-methylbenzamide in ethanol using sodium bicarbonate.
- Isolate the intermediate 2-(2-chloro-5-trifluoromethylpyrimidin-4-ylamino)-N-methylbenzamide via filtration.
- Conduct Buchwald-Hartwig amination with various amines using Pd(OAc)2, xantphos, and NaOtBu in 1,4-dioxane at 100°C.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible benefits beyond mere chemical elegance. The reliance on commodity chemicals such as 2,4-dichloro-5-trifluoromethylpyrimidine and simple benzamides ensures a stable and resilient supply chain, mitigating the risks associated with sourcing exotic or single-source raw materials. The modular nature of the synthesis allows for the production of multiple analogues from a single batch of the common intermediate, significantly enhancing inventory flexibility and reducing the capital tied up in specialized stock. Furthermore, the elimination of cryogenic conditions and the use of standard heating protocols simplify the engineering requirements for scale-up, making the transition from gram-scale laboratory synthesis to multi-kilogram pilot production seamless and predictable.
- Cost Reduction in Manufacturing: The streamlined two-step process eliminates the need for protecting group strategies often required in traditional pyrimidine synthesis, thereby reducing the number of unit operations and the consumption of auxiliary reagents. By avoiding expensive custom building blocks and utilizing a catalytic amount of palladium which can potentially be recovered, the overall material costs are significantly optimized. The high selectivity of the reaction minimizes the formation of difficult-to-remove impurities, lowering the cost associated with chromatographic purification and increasing the overall throughput of the manufacturing facility.
- Enhanced Supply Chain Reliability: The robustness of the chemistry against minor variations in reaction conditions translates to higher batch-to-batch consistency, a critical factor for regulatory compliance. Since the key starting materials are produced by multiple global suppliers, the risk of supply disruption is minimized, ensuring continuous availability of the intermediate for downstream drug substance manufacturing. This reliability allows for better production planning and shorter lead times, enabling pharmaceutical partners to accelerate their development timelines without the fear of raw material shortages.
- Scalability and Environmental Compliance: The process utilizes solvents like ethanol and 1,4-dioxane, which are well-understood in terms of waste management and recovery, facilitating adherence to environmental regulations. The absence of hazardous reagents such as strong acids or pyrophoric organometallics simplifies the safety profile of the plant operations. Moreover, the high atom economy of the coupling reaction reduces the volume of chemical waste generated per kilogram of product, aligning with green chemistry principles and reducing the environmental footprint of the manufacturing process.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is vital for effective collaboration between CDMOs and pharmaceutical innovators. The following questions address common inquiries regarding the scalability, purity, and application of these 5-trifluoromethylpyrimidine derivatives. Our technical team has compiled these insights based on the patent data and our internal expertise in heterocyclic chemistry to provide clarity on the implementation of this technology.
Q: What is the key advantage of this synthetic route for 5-trifluoromethylpyrimidines?
A: The route utilizes a highly regioselective two-step process that allows for the late-stage diversification of the 2-position amine, enabling rapid SAR exploration without redesigning the core scaffold.
Q: How is impurity control managed during the amination step?
A: By using a robust Pd/Xantphos catalyst system under inert atmosphere, the reaction minimizes dehalogenation byproducts and ensures high conversion of the chloro-intermediate to the desired secondary amine.
Q: Are the starting materials commercially available for scale-up?
A: Yes, the primary building block, 2,4-dichloro-5-trifluoromethylpyrimidine, is a commodity chemical, ensuring supply chain stability and cost-effectiveness for large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethylpyrimidine Derivative Supplier
At NINGBO INNO PHARMCHEM, we combine deep scientific expertise with industrial capability to bring complex molecules from the lab to the market. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and efficiency. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 5-trifluoromethylpyrimidine derivative meets the highest international standards. We understand the critical nature of oncology drug development and are committed to being a partner that delivers both quality and reliability.
We invite you to engage with our technical procurement team to discuss how this novel synthetic route can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain valuable insights into the economic benefits of adopting this methodology for your specific program. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project requirements, ensuring a smooth and successful path forward for your anti-tumor drug candidates.