Scalable Manufacturing of Tetraisobutyryl Nucleoside Analogs for Antiviral APIs
Scalable Manufacturing of Tetraisobutyryl Nucleoside Analogs for Antiviral APIs
The global demand for effective antiviral therapeutics has intensified the search for robust, scalable synthetic routes for key pharmaceutical intermediates. Patent CN114573590A introduces a groundbreaking preparation method for tetraisobutyryl nucleoside analogs, which serve as critical precursors for potent antiviral agents like VV116 and A131. These compounds have demonstrated significant inhibitory activity against SARS-CoV-2, respiratory syncytial virus, and hepatitis C virus. The disclosed technology addresses long-standing challenges in nucleoside chemistry by offering a pathway that is not only high-yielding but also operationally simple, avoiding the pitfalls of traditional multi-step protection strategies. This innovation represents a pivotal shift towards more sustainable and cost-efficient manufacturing of complex nucleoside derivatives.
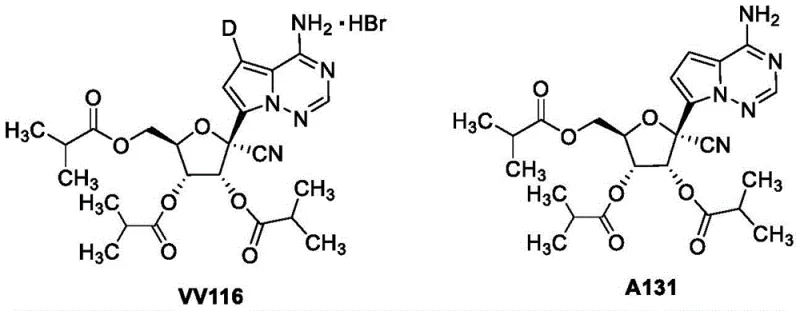
For R&D directors and process chemists, the structural integrity and purity of these intermediates are paramount. The patent details a direct acylation strategy that modifies the parent nucleoside at multiple positions simultaneously, creating a tetra-acylated species that can be selectively deprotected to the desired tri-isobutyrate prodrug form. This approach significantly streamlines the synthesis of VV116, a molecule where the triisobutyrate prodrug form drastically improves physicochemical properties and in vivo metabolic stability compared to the parent nucleoside. By understanding the nuances of this new methodology, pharmaceutical manufacturers can better assess the feasibility of integrating these intermediates into their existing supply chains for antiviral drug production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of triisobutyrate prodrugs like those found in VV116 has been plagued by inefficiencies and operational complexities. Existing literature, such as reports in Cell Research (2021), relied heavily on protecting group strategies. These traditional routes necessitate additional reaction steps to install and subsequently remove protecting groups, which inherently lowers the overall yield and increases the generation of chemical waste. Furthermore, other documented methods, such as those published in J. Med. Chem. (2021), utilized condensation agents like diisopropylcarbodiimide (DIC) in polar aprotic solvents like DMF. While effective on a small scale, these reagents generate substantial amounts of urea by-products that are notoriously difficult to remove, complicating purification and posing risks to product purity.
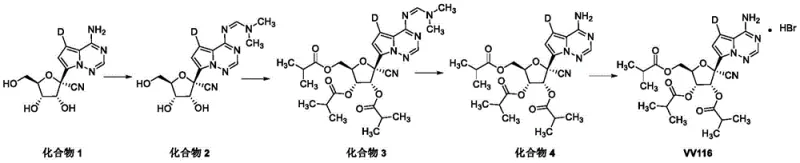
Another significant bottleneck identified in prior art, specifically patent CN113735862, involved the direct reaction of the parent nucleoside with isobutyric anhydride. Unfortunately, this method suffered from abysmal conversion rates, with reported yields as low as 35%. Such low efficiency is commercially untenable for large-scale API manufacturing, where material costs and throughput are critical drivers of profitability. The accumulation of unreacted starting materials and side products in these conventional processes necessitates extensive chromatographic purification, which is neither economically nor environmentally sustainable for industrial applications. These limitations underscore the urgent need for a more direct, high-yielding synthetic alternative.
The Novel Approach
The methodology outlined in CN114573590A offers a transformative solution by employing a direct acylation protocol that bypasses the need for orthogonal protecting groups. The core innovation lies in the reaction of a compound of Formula II with an acylating agent, such as isobutyryl chloride or isobutyric anhydride, under the influence of a specific base system. This single-step transformation efficiently installs four isobutyryl groups onto the nucleoside scaffold. The process is characterized by mild reaction conditions, typically operating between 35°C and 45°C, which minimizes thermal degradation of the sensitive nucleoside core. By optimizing the molar ratios of the acylating agent and base to approximately 1:4.5-5.0, the reaction achieves near-quantitative conversion, resulting in isolated yields consistently exceeding 80%, and in some examples reaching up to 91%.
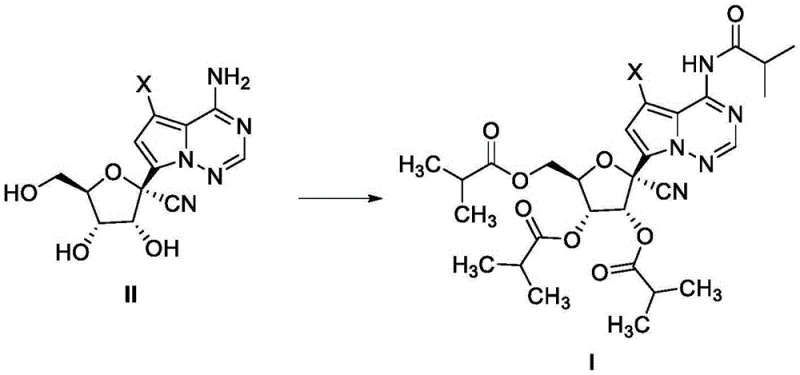
This novel approach not only enhances yield but also simplifies the downstream processing. The workup procedure involves a straightforward aqueous quench followed by extraction and crystallization, eliminating the need for column chromatography. The ability to purify the product via crystallization from heptane is a major advantage for scale-up, as it ensures high purity without the solvent intensity associated with chromatographic methods. Furthermore, the versatility of this method allows for the synthesis of various halogenated and deuterated analogs, providing a flexible platform for developing next-generation antiviral candidates with improved pharmacokinetic profiles.
Mechanistic Insights into Base-Catalyzed Multi-Site Acylation
The success of this tetra-acylation reaction hinges on the precise selection of the base and solvent system to manage the reactivity of the multiple hydroxyl and amine functionalities present on the nucleoside. The patent specifies the use of tertiary amines like triethylamine or nucleophilic catalysts like 4-dimethylaminopyridine (DMAP). Triethylamine serves as a proton scavenger, neutralizing the hydrochloric acid generated during the reaction with isobutyryl chloride, thereby driving the equilibrium towards product formation. DMAP acts as a potent acylation catalyst, forming a reactive acylpyridinium intermediate that is more electrophilic than the acid chloride itself. This activation is crucial for acylating the sterically hindered secondary hydroxyl groups on the ribose ring and the exocyclic amine on the heterocyclic base.
Impurity control is meticulously managed through temperature regulation and stoichiometry. Operating at lower temperatures (0°C) during the addition of the acylating agent prevents exothermic runaway and minimizes side reactions such as ester migration or hydrolysis. Subsequent warming to 35-45°C ensures complete conversion of the starting material. The choice of dichloromethane (DCM) as the preferred solvent provides an optimal balance of solubility for both the polar nucleoside starting material and the organic reagents, while facilitating easy phase separation during the aqueous workup. This mechanistic understanding allows process chemists to fine-tune the reaction parameters to suppress specific impurities, ensuring the final intermediate meets the stringent quality standards required for pharmaceutical applications.
How to Synthesize Tetraisobutyryl Nucleoside Analogs Efficiently
The synthesis of these high-value intermediates is designed for operational simplicity and robustness. The process begins by dissolving the nucleoside starting material in dichloromethane and adding the base system under controlled cooling. The acylating agent is then introduced dropwise to maintain thermal stability. Following the reaction period, the mixture is quenched into ice water, and the organic layer is washed with saturated sodium bicarbonate to remove acidic by-products. The crude product is concentrated and precipitated using non-polar solvents like n-heptane, yielding a solid that can be directly used in subsequent steps or further purified if necessary. Detailed standardized synthesis steps are provided in the guide below.
- React the nucleoside compound (Formula II) with isobutyryl chloride or anhydride in dichloromethane using triethylamine and DMAP as bases at 35-45°C.
- Quench the reaction with ice water, wash with saturated sodium bicarbonate, and concentrate the organic phase.
- Precipitate the product using n-heptane, filter, and dry to obtain the tetraisobutyryl intermediate with yields exceeding 80%.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this new synthetic route offers tangible benefits that extend beyond mere chemical yield. The elimination of expensive and hazardous condensing agents like DIC significantly reduces the raw material cost per kilogram of the intermediate. Moreover, the avoidance of DMF, a solvent with strict regulatory limits due to its reproductive toxicity, simplifies environmental compliance and waste disposal protocols. The process relies on common, commodity chemicals such as triethylamine, isobutyryl chloride, and dichloromethane, which are readily available in the global market, ensuring supply chain continuity and reducing the risk of vendor lock-in for specialized reagents.
- Cost Reduction in Manufacturing: The dramatic improvement in yield from roughly 35% in prior art to over 80% in this new process effectively halves the consumption of the expensive nucleoside starting material per unit of output. This material efficiency translates directly into a lower cost of goods sold (COGS). Additionally, the simplified workup procedure, which replaces time-consuming chromatography with crystallization, reduces labor hours, solvent consumption, and equipment occupancy time. The removal of heavy metal catalysts in the initial acylation step also negates the need for costly metal scavenging resins and the associated analytical testing for residual metals, further driving down operational expenses.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, which tolerate a range of temperatures and use stable reagents, makes the process less susceptible to batch failures. High reproducibility is critical for maintaining consistent supply to downstream API manufacturers. The ability to produce deuterated analogs using the same platform technology adds value for clients seeking isotopically labeled standards or drugs with improved metabolic stability. Since the raw materials are bulk commodities, procurement teams can leverage competitive bidding among multiple suppliers, mitigating the risk of supply disruptions that often plague specialty chemical markets.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated in multi-gram to decagram scales in the patent examples with consistent results. The use of dichloromethane, while requiring proper handling, allows for efficient solvent recovery and recycling, aligning with green chemistry principles when managed correctly. The reduction in by-product formation means less chemical waste is generated per kilogram of product, lowering the environmental footprint of the manufacturing site. This alignment with sustainability goals is increasingly important for pharmaceutical companies aiming to meet corporate social responsibility targets and regulatory expectations for green manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These answers are derived directly from the experimental data and claims presented in CN114573590A, providing a reliable basis for decision-making. Understanding these details helps stakeholders evaluate the fit of this technology within their specific production contexts and quality frameworks.
Q: What is the primary advantage of the new acylation method over prior art?
A: The new method eliminates complex protecting group strategies and avoids toxic condensing agents like DCC, achieving yields over 80% compared to previous 35% yields.
Q: Can this process be adapted for deuterated analogs?
A: Yes, the patent describes a catalytic dehalogenation step using Pd/C and deuterium gas to introduce deuterium labels efficiently.
Q: Is the process suitable for large-scale industrial production?
A: Absolutely. The process uses mild temperatures (35-45°C), common solvents like DCM, and simple aqueous workups, making it highly scalable.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable VV116 Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of high-quality intermediates in the race to develop effective antiviral therapies. Our technical team has thoroughly analyzed the pathways described in CN114573590A and is fully prepared to execute this advanced synthesis at scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of tetraisobutyryl nucleoside analogs meets the highest industry standards for potency and impurity profiles.
We invite you to collaborate with us to optimize your supply chain for VV116 and related antiviral programs. Our experts can provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this new process can impact your bottom line. We encourage potential partners to contact our technical procurement team to request specific COA data and route feasibility assessments. Let us be your trusted partner in navigating the complexities of nucleoside chemistry and delivering the building blocks necessary for the next generation of life-saving medicines.