Advanced Synthesis of Safer Gastrointestinal Prokinetic Intermediates for Commercial API Production
Advanced Synthesis of Safer Gastrointestinal Prokinetic Intermediates for Commercial API Production
The pharmaceutical landscape for gastrointestinal motility disorders has long been dominated by compounds that balance efficacy against significant cardiovascular and neurological risks. Patent CN1653063A introduces a breakthrough in this domain by disclosing the synthesis and pharmacological profile of (S)-4-amino-5-chloro-2-methoxy-N-[1-[1-(2-tetrahydrofurylcarbonyl)-4-piperidinylmethyl]-4-piperidinyl]benzamide. This specific stereoisomer represents a critical evolution in 5-HT4 receptor agonist design, offering potent prokinetic activity with a markedly improved safety profile regarding QTc interval prolongation and dopamine D2 receptor antagonism. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediate suppliers, understanding the nuanced chemistry behind this molecule is essential for securing a stable supply of next-generation gastrointestinal therapeutics. The patent details a robust synthetic pathway that leverages standard amide coupling techniques while emphasizing the importance of stereochemical integrity at the tetrahydrofuran moiety.
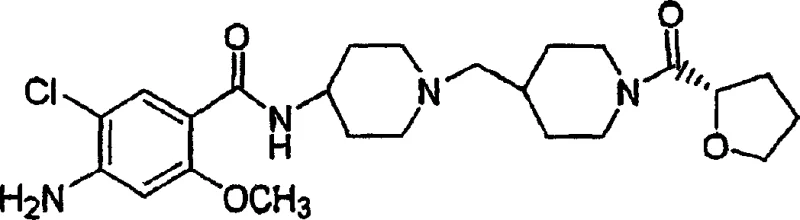
This compound serves as a high-value pharmaceutical intermediate, bridging the gap between early-stage discovery and commercial API manufacturing. The structural complexity, involving a bis-piperidine linker and a chiral heterocyclic acyl group, demands precise process control to ensure batch-to-batch consistency. By targeting the specific (S)-enantiomer, the invention addresses the historical limitations of earlier prokinetic agents like cisapride, which were withdrawn or restricted due to cardiac arrhythmias. Consequently, the demand for high-purity precursors and intermediates capable of delivering this specific stereochemistry is rising within the global supply chain for gastroenterology medications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art, such as JP-A-2000-80081, disclosed a broad genus of benzamide derivatives where the acyl substituent could vary widely, including furan and tetrahydropyran rings. However, these earlier iterations often suffered from a lack of selectivity, leading to off-target binding at dopamine D2 receptors which causes extrapyramidal symptoms, or at cardiac ion channels leading to QTc prolongation. Specifically, the racemic mixtures or the (R)-isomers described in previous literature demonstrated significantly higher affinity for dopamine receptors, negating the safety benefits sought in modern prokinetic therapy. Furthermore, conventional synthesis routes for similar bis-piperidine scaffolds often relied on non-selective alkylation or acylation steps that produced difficult-to-separate impurities, complicating the purification process and driving up manufacturing costs. The inability to consistently produce the specific (S)-tetrahydrofuran-2-carbonyl motif with high optical purity was a major bottleneck in developing safer analogs.
The Novel Approach
The novel approach detailed in CN1653063A overcomes these hurdles by strictly defining the stereochemistry at the 2-position of the tetrahydrofuran ring. By utilizing (S)-tetrahydrofuran-2-carboxylic acid as a key building block, the process ensures that the final amide product possesses the requisite spatial configuration to act as a selective 5-HT4 agonist. This method diverges from prior art by demonstrating that the (S)-configuration drastically reduces affinity for dopamine D2 receptors (IC50 > 10000 nM) while maintaining potent agonistic activity at 5-HT4 receptors (IC50 13.5 nM). From a manufacturing perspective, this specificity allows for the implementation of targeted crystallization strategies, such as forming the fumarate salt, to upgrade optical purity to greater than 99% ee without resorting to expensive preparative chiral HPLC. This shift from separation-heavy processes to stereocontrolled synthesis represents a significant advancement in cost reduction in API manufacturing for complex gastrointestinal agents.
Mechanistic Insights into Stereoselective Amide Coupling
The core chemical transformation in this patent revolves around the formation of two distinct amide bonds within a highly functionalized scaffold. The first critical mechanistic step involves the acylation of the secondary amine on the piperidine ring with (S)-tetrahydrofuran-2-carboxylic acid. This reaction typically employs carbodiimide-based condensing agents, such as EDC·HCl or DCC, often in the presence of additives like HOBt to suppress racemization of the chiral alpha-center. The mechanism proceeds through the formation of an O-acylisourea intermediate, which is then attacked by the nucleophilic piperidine nitrogen. Maintaining mild reaction temperatures, generally between -10°C to 25°C, is crucial to prevent epimerization of the sensitive chiral center adjacent to the carbonyl group. The choice of solvent, such as chloroform or dichloromethane, facilitates the solubility of the polar intermediates while allowing for easy workup and isolation of the coupled product.
Following the installation of the chiral acyl group, the second amide bond is formed with the benzoic acid moiety. This step requires careful management of the free amino group on the benzene ring, which can compete for acylation if not properly managed or if the reactivity ratios are not optimized. The patent suggests that the steric hindrance provided by the bulky bis-piperidine-tetrahydrofuran intermediate favors the reaction at the intended site when using activated derivatives of 4-amino-5-chloro-2-methoxybenzoic acid. Impurity control is achieved through the strategic use of protecting groups (L1 and L2) during the assembly of the piperidine linker, as illustrated in the synthesis of intermediate (V). These protecting groups, such as benzyloxycarbonyl or tert-butoxycarbonyl, are orthogonal, allowing for sequential deprotection that minimizes side reactions and ensures the final product meets stringent purity specifications required for clinical applications.
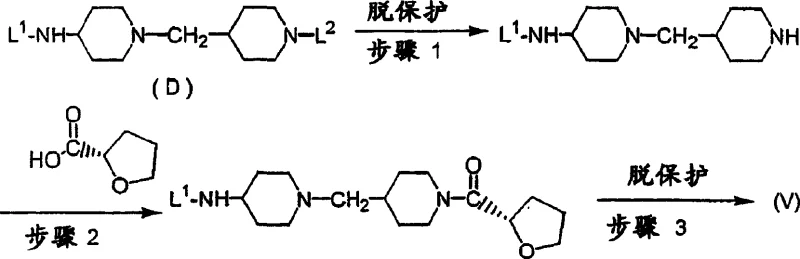
How to Synthesize (S)-Tetrahydrofuran Benzamide Derivatives Efficiently
The synthesis of this high-value intermediate follows a logical progression designed to maximize yield and optical purity while minimizing waste. The process begins with the preparation of the chiral piperidine scaffold, where protecting groups are strategically removed to expose the reactive amine. This is followed by the coupling with the chiral acid and finally the attachment of the benzamide head group. Each step is optimized for scalability, utilizing reagents that are readily available in the fine chemical market. For a detailed breakdown of the specific reaction conditions, stoichiometry, and workup procedures, please refer to the standardized guide below which outlines the critical operational parameters for successful execution.
- Deprotection of the bis-protected piperidine scaffold (L1/L2 groups) using hydrogenolysis or hydrolysis to reveal the reactive amine sites.
- Stereoselective acylation of the secondary piperidine nitrogen using (S)-tetrahydrofuran-2-carboxylic acid activated by carbodiimide condensing agents.
- Final amide bond formation between the functionalized piperidine intermediate and 4-amino-5-chloro-2-methoxybenzoic acid derivatives under controlled pH conditions.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the synthesis route described in CN1653063A offers tangible benefits beyond mere technical feasibility. The primary advantage lies in the robustness of the supply chain for raw materials. Key starting materials like (S)-tetrahydrofuran-2-carboxylic acid and substituted piperidines are commodity chemicals produced by multiple global vendors, reducing the risk of single-source bottlenecks. This diversity in sourcing options enhances supply chain reliability and provides leverage in price negotiations, ensuring that cost reduction in pharmaceutical intermediate manufacturing is achievable without compromising quality. Furthermore, the avoidance of precious metal catalysts or exotic reagents simplifies the regulatory filing process and reduces the environmental footprint associated with heavy metal removal and disposal.
- Cost Reduction in Manufacturing: The process eliminates the need for expensive chiral resolution steps typically required for racemic mixtures. By starting with enantiomerically pure (S)-acid and maintaining stereochemical integrity throughout the coupling, manufacturers can avoid the significant yield losses associated with recrystallization or chromatographic separation of enantiomers. Additionally, the ability to purify the final product via simple salt formation (e.g., fumarate or maleate salts) rather than complex column chromatography significantly lowers solvent consumption and processing time. This streamlined downstream processing translates directly into lower operating expenses and a more competitive cost structure for the final API.
- Enhanced Supply Chain Reliability: The synthetic route relies on stable, shelf-stable reagents that do not require specialized storage conditions like cryogenic temperatures or inert atmospheres beyond standard nitrogen blanketing. This simplicity reduces the logistical complexity of transporting and storing hazardous or unstable intermediates. Moreover, the reaction conditions are tolerant of minor variations in temperature and mixing rates, making the process robust for transfer between different manufacturing sites or contract manufacturing organizations (CMOs). This flexibility ensures continuity of supply even in the face of regional disruptions or equipment maintenance schedules.
- Scalability and Environmental Compliance: The chemistry described is inherently scalable from kilogram to multi-ton production scales without fundamental changes to the reaction mechanism. The use of common organic solvents like ethyl acetate, ethanol, and chloroform allows for established recovery and recycling protocols, aligning with green chemistry principles and reducing waste disposal costs. The absence of genotoxic reagents or highly toxic catalysts simplifies the control strategy for impurities, facilitating faster regulatory approval and market entry. This environmental compliance is increasingly critical for maintaining a social license to operate in strict regulatory jurisdictions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this specific gastrointestinal intermediate. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making.
Q: Why is the (S)-stereochemistry critical for this gastrointestinal agent?
A: The (S)-configuration at the tetrahydrofuran ring is essential for maintaining high affinity for 5-HT4 receptors while minimizing binding to dopamine D2 receptors, thereby reducing central nervous system side effects compared to the racemic mixture or (R)-isomer.
Q: How does this synthesis route improve supply chain stability?
A: The process utilizes commercially available starting materials like (S)-tetrahydrofuran-2-carboxylic acid and avoids rare transition metal catalysts, ensuring consistent raw material availability and reducing dependency on specialized catalytic supply chains.
Q: What are the purity specifications for the final intermediate?
A: The patent describes achieving optical purity greater than 99% ee through crystallization of the fumarate salt, meeting stringent requirements for chiral pharmaceutical intermediates without needing complex chiral chromatography at scale.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (S)-Tetrahydrofuran Benzamide Supplier
As the global demand for safer gastrointestinal therapies continues to grow, partnering with an experienced CDMO is vital for translating patent chemistry into commercial reality. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move seamlessly from pilot plant to full-scale manufacturing. Our facility is equipped with rigorous QC labs and advanced analytical instrumentation capable of verifying stringent purity specifications, including chiral purity assessment via HPLC, to guarantee that every batch meets the high standards required for pharmaceutical intermediates.
We invite you to engage with our technical procurement team to discuss your specific requirements for this compound or related analogs. By requesting a Customized Cost-Saving Analysis, you can gain insights into how our optimized processes can reduce your overall cost of goods. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing us to demonstrate our commitment to quality, speed, and technical excellence in the supply of complex pharmaceutical building blocks.