Scalable Synthesis of Chiral Pyrrolidine-1H-Tetrazole Derivatives for Advanced Pharmaceutical Applications
The pharmaceutical and fine chemical industries continuously seek robust methodologies for constructing nitrogen-rich heterocycles, particularly tetrazoles, which serve as critical bioisosteres for carboxylic acids and essential motifs in modern drug design. Patent CN101228153A introduces a transformative approach for the preparation of (S)-pyrrolidine-1H-tetrazole derivatives, addressing long-standing safety and purity challenges associated with traditional synthesis routes. This technology enables the production of high-purity organocatalysts and active pharmaceutical ingredient (API) intermediates by utilizing dialkylaluminum or dialkylboron azides as the key cyclization reagents. Unlike conventional methods that rely on toxic organotin compounds or thermally unstable ammonium azides, this novel process ensures a cleaner reaction profile with significantly reduced ecological impact. For R&D directors and process chemists, this represents a pivotal shift towards greener chemistry without compromising the stereochemical integrity required for chiral drug synthesis. The ability to access these derivatives in racemic or enantiomerically pure forms (>99% ee) opens new avenues for developing asymmetric catalysts used in aldol, Mannich, and Michael reactions.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 5-substituted-1H-tetrazoles has been plagued by significant safety and environmental hurdles, primarily due to the reliance on organotin azides such as trimethyltin azide. While these reagents can effect the transformation of nitriles to tetrazoles, they introduce severe toxicity concerns that complicate downstream processing and waste management. The removal of tin residues often requires expensive scavenging agents or complex chromatographic separations, which drastically increases the cost of goods sold (COGS) and extends production lead times. Furthermore, alternative methods utilizing trialkylammonium or tetraalkylammonium azides pose substantial explosion risks; these salts can form volatile sublimates at elevated reactor temperatures, creating hazardous conditions that are difficult to mitigate on a commercial scale. Consequently, manufacturers face stringent regulatory scrutiny and higher insurance premiums when employing these legacy technologies, limiting their ability to scale production efficiently for global supply chains.
The Novel Approach
The methodology disclosed in CN101228153A circumvents these critical bottlenecks by employing dialkylaluminum azides (e.g., diethylaluminum azide) or dialkylboron azides generated in situ from readily available precursors. This innovation allows the cycloaddition reaction between the nitrile group of the pyrrolidine substrate and the azide moiety to proceed under much milder and safer conditions, typically ranging from 40°C to 120°C in inert hydrocarbon solvents like xylene or toluene. By eliminating heavy metals from the reagent system, the resulting crude product exhibits a vastly superior impurity profile, facilitating simpler crystallization or extraction protocols. This approach not only enhances operator safety by removing explosion hazards associated with ammonium azides but also aligns with modern green chemistry principles by reducing the E-factor of the synthesis. For procurement managers, this translates to a more reliable supply chain with fewer disruptions caused by hazardous material handling regulations and waste disposal constraints.
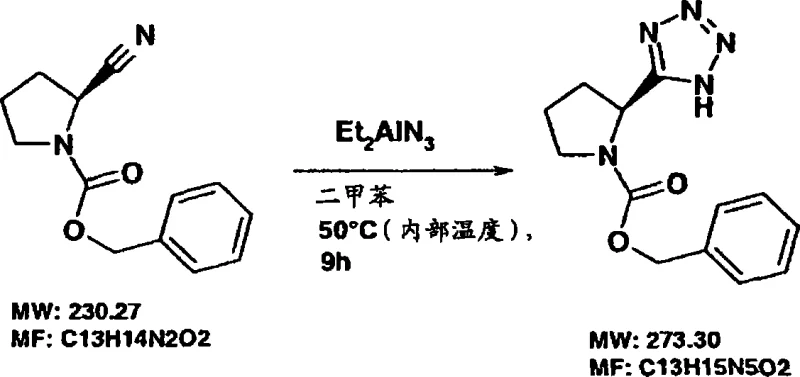
Mechanistic Insights into Organoaluminum Azide Cyclization
The core of this technological advancement lies in the unique reactivity of the aluminum-nitrogen bond within the dialkylaluminum azide species. When the protected pyrrolidine-2-carbonitrile (Formula IIa) interacts with the azide reagent (Formula IIb), the aluminum center acts as a Lewis acid, activating the nitrile triple bond towards nucleophilic attack by the terminal nitrogen of the azide group. This coordination facilitates a [2+3] dipolar cycloaddition that constructs the tetrazole ring with high regioselectivity. Crucially, the aluminum atom remains coordinated to the newly formed tetrazole nitrogen, stabilizing the intermediate and preventing side reactions that often plague uncatalyzed thermal cycloadditions. The process tolerates a wide array of substituents (R1, R2, R3) on the pyrrolidine ring, including alkyl, aryl, and halogen groups, provided they do not interfere with the azide functionality. Following the cyclization, a carefully controlled acidic or basic workup cleaves the aluminum-tetrazole bond, releasing the free tetrazole or its salt form. This mechanism ensures that the chiral center at the 2-position of the pyrrolidine ring is preserved, maintaining the optical purity essential for subsequent applications in asymmetric organocatalysis.
Impurity control is inherently built into this mechanistic pathway due to the specificity of the aluminum-mediated activation. Unlike tin-based reagents that may promote non-specific decomposition or transmetallation side products, the organoaluminum system favors the desired cyclization exclusively. The patent data indicates that by adjusting the molar ratio of azide to nitrile (optimally between 1.2:1 and 1.8:1), manufacturers can drive the reaction to completion while minimizing the formation of bis-tetrazole byproducts or oligomers. Furthermore, the choice of protecting group (Z1) on the pyrrolidine nitrogen, such as Cbz or Boc, plays a vital role in solubility and crystallization behavior during the isolation phase. The robustness of these protecting groups under the reaction conditions (up to 120°C) ensures that the amine functionality remains masked until the final deprotection step, thereby preventing polymerization or self-condensation of the reactive amine. This level of control over the reaction trajectory is paramount for achieving the >99% ee specifications required by top-tier pharmaceutical clients.
How to Synthesize (S)-Pyrrolidine-1H-Tetrazole Derivatives Efficiently
The practical implementation of this synthesis route involves a streamlined sequence that begins with the in situ generation of the azide reagent, followed by the addition of the nitrile substrate and a controlled thermal cycle. Detailed operational parameters, such as the specific temperature ramps and quenching protocols described in the patent examples, are critical for maximizing yield and safety. Operators must adhere to strict anhydrous conditions during the initial mixing phase to prevent premature hydrolysis of the sensitive aluminum reagent. The subsequent workup involves a pH-controlled extraction strategy that effectively separates the organic tetrazole product from inorganic salts and aluminum residues. For a comprehensive understanding of the precise stoichiometry and equipment requirements needed to replicate these results in a pilot plant setting, please refer to the standardized synthesis guide below.
- Preparation of the azide reagent by reacting granular sodium azide with dialkylaluminum chloride (e.g., diethylaluminum chloride) in an inert solvent like xylene or toluene at 0°C to room temperature.
- Addition of the protected pyrrolidine-2-carbonitrile starting material (e.g., Cbz or Boc protected) to the azide solution, followed by heating to 40°C-80°C to facilitate the [2+3] cycloaddition reaction.
- Quenching the reaction mixture with aqueous acid or base to decompose excess azide, followed by extraction, purification, and optional deprotection (e.g., hydrogenation) to yield the final tetrazole product.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this organoaluminum-based synthesis route offers profound strategic benefits for supply chain stability and cost management, primarily driven by the elimination of hazardous reagents and simplified purification workflows. By removing the need for organotin compounds, manufacturers can bypass the expensive and time-consuming heavy metal clearance steps that typically dominate the downstream processing of tetrazole intermediates. This reduction in unit operations directly correlates to lower utility consumption, reduced solvent usage, and decreased labor hours per kilogram of product. Furthermore, the enhanced thermal safety profile of the dialkylaluminum azide method mitigates the risk of batch loss due to runaway exotherms or decomposition events, ensuring higher overall equipment effectiveness (OEE) and more predictable delivery schedules. These factors collectively contribute to a more resilient supply chain capable of meeting the rigorous demands of Just-In-Time manufacturing environments without compromising on quality or compliance standards.
- Cost Reduction in Manufacturing: The transition away from toxic tin reagents eliminates the necessity for specialized waste treatment facilities and costly metal scavenging resins, resulting in substantial operational expenditure savings. Additionally, the use of commodity chemicals like sodium azide and dialkylaluminum chlorides as starting materials ensures a stable and competitive raw material pricing structure, shielding the production budget from volatility associated with specialty organometallic reagents. The simplified isolation procedure, often relying on straightforward crystallization rather than complex chromatography, further drives down the cost of goods by reducing solvent recovery loads and increasing throughput capacity.
- Enhanced Supply Chain Reliability: Sourcing of key reagents for this process is significantly more robust compared to legacy methods, as dialkylaluminum chlorides and sodium azide are widely available from multiple global suppliers, reducing single-source dependency risks. The improved safety profile of the reaction mixture also facilitates easier transportation and storage of intermediates, minimizing logistical delays caused by hazardous material shipping restrictions. This reliability allows procurement teams to negotiate better long-term contracts and maintain optimal inventory levels, ensuring continuous availability of critical chiral intermediates for downstream API synthesis without unexpected interruptions.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing standard stainless steel reactors and common hydrocarbon solvents that are compatible with existing infrastructure in most multipurpose chemical plants. The absence of persistent organic pollutants like organotins simplifies environmental permitting and reduces the regulatory burden associated with effluent discharge, making it easier to expand production capacity in regions with strict ecological laws. This alignment with sustainability goals not only future-proofs the manufacturing asset but also enhances the brand reputation of the supply partner among environmentally conscious pharmaceutical customers seeking green chemistry solutions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this tetrazole synthesis technology, drawing directly from the experimental data and scope defined in the patent documentation. Understanding these nuances is essential for process engineers and quality assurance teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The answers provided reflect the specific capabilities and limitations observed during the development of the organoaluminum azide methodology, ensuring that expectations regarding yield, purity, and safety are grounded in empirical evidence.
Q: Why is the organoaluminum azide method preferred over organotin azides for tetrazole synthesis?
A: Organotin azides are highly toxic and create significant ecological problems, requiring extensive and costly wastewater recycling processes to remove tin residues. The organoaluminum method described in CN101228153A eliminates heavy metal contamination, simplifying purification and reducing environmental compliance costs.
Q: What protecting groups are compatible with this tetrazole formation process?
A: The process is compatible with standard amino protecting groups used in peptide chemistry, including benzyloxycarbonyl (Cbz), tert-butoxycarbonyl (Boc), benzyl (Bn), and silyl groups. These groups remain stable during the azide cyclization and can be removed subsequently under standard acidic or hydrogenation conditions.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the method avoids the explosion risks associated with volatile ammonium azide sublimates at high temperatures. By using dialkylaluminum azides in high-boiling solvents like xylene at moderate temperatures (50°C-120°C), the process offers improved thermal safety and scalability for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pyrrolidine-1H-Tetrazole Derivative Supplier
NINGBO INNO PHARMCHEM stands at the forefront of translating advanced patent technologies like CN101228153A into commercial reality, offering unparalleled expertise in the scale-up of complex chiral intermediates. Our facility is equipped with state-of-the-art reactors and rigorous QC labs capable of handling sensitive organometallic reactions under strictly controlled anhydrous conditions, ensuring consistent batch-to-batch reproducibility. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, adhering to stringent purity specifications required by global regulatory bodies. Our commitment to process safety and environmental stewardship means that every kilogram of pyrrolidine-tetrazole derivative we produce meets the highest standards of quality while minimizing ecological impact, providing our partners with a secure and sustainable source of critical building blocks.
We invite potential collaborators to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain detailed insights into the economic advantages of switching to this tin-free methodology for your supply chain. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that optimize both your R&D timelines and commercial manufacturing costs. Let us be your trusted partner in navigating the complexities of modern pharmaceutical intermediate synthesis.