Revolutionizing Vilanterol Intermediate Production: A Scalable Crystallization Strategy for Global API Supply Chains
The pharmaceutical landscape for respiratory therapies continues to evolve, driven by the demand for high-purity active pharmaceutical ingredients (APIs) such as Vilanterol, a potent long-acting β2-receptor agonist. A pivotal development in this sector is documented in patent CN115286491A, which discloses a groundbreaking preparation method for the key intermediate 2-[2-(6-bromohexyloxy)ethoxymethyl]-1,3-dichlorobenzene. This technical disclosure addresses critical bottlenecks in the existing supply chain by introducing a route that transforms an traditionally oily, difficult-to-purify intermediate into a crystalline solid amenable to standard industrial purification techniques. For R&D directors and supply chain strategists, this shift represents a move away from laboratory-scale constraints toward robust, commercial-grade manufacturability. The patent outlines a multi-step synthesis starting from inexpensive 2,6-dichlorobenzyl alcohol, leveraging sequential nucleophilic substitutions and a strategic sulfonate esterification to achieve product purity exceeding 99%. This report analyzes the technical merits of this innovation, evaluating its potential to redefine cost structures and reliability metrics for global procurement teams seeking reliable pharmaceutical intermediates supplier partnerships.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of this critical Vilanterol precursor has been plagued by significant operational inefficiencies that hinder large-scale production. Conventional literature and prior art typically employ 2,6-dichlorobenzyl bromide as the starting material, subjecting it to two-step nucleophilic substitution reactions to construct the necessary ether backbone. While chemically feasible on a small scale, this approach suffers from severe drawbacks when translated to industrial volumes. The primary issue lies in the physical state of the intermediates and the final product; they exist as viscous oils with high boiling points, rendering them impossible to purify through standard crystallization techniques. Consequently, manufacturers are forced to rely on column chromatography, a process that is notoriously solvent-intensive, time-consuming, and difficult to automate or scale continuously. 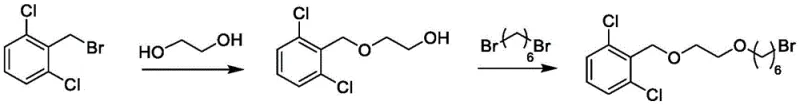 Furthermore, the use of benzyl bromide introduces stability concerns and higher raw material costs compared to alcohol counterparts. The accumulation of by-products in these oily phases complicates the impurity profile, often necessitating repeated purification cycles that erode overall yield and extend lead times. For a procurement manager, these factors translate into unpredictable supply continuity and inflated cost bases, making the conventional route unsustainable for meeting the rigorous demands of modern respiratory drug manufacturing.
Furthermore, the use of benzyl bromide introduces stability concerns and higher raw material costs compared to alcohol counterparts. The accumulation of by-products in these oily phases complicates the impurity profile, often necessitating repeated purification cycles that erode overall yield and extend lead times. For a procurement manager, these factors translate into unpredictable supply continuity and inflated cost bases, making the conventional route unsustainable for meeting the rigorous demands of modern respiratory drug manufacturing.
The Novel Approach
In stark contrast, the methodology presented in CN115286491A introduces a paradigm shift by utilizing 2,6-dichlorobenzyl alcohol as the foundational building block. This strategic change in starting material initiates a cascade of reactions designed to introduce a crystallizable checkpoint within the synthesis sequence. The novel route involves the formation of a sulfonate ester intermediate (Formula II), which, unlike its predecessors in the old route, manifests as a white solid. This physical transformation is the linchpin of the entire process improvement. 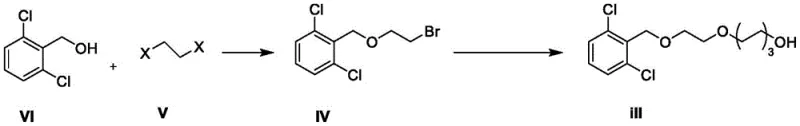 By converting the terminal hydroxyl group into a mesylate, the process creates a molecule with distinct lattice energy properties that allow for purification via simple solvent recrystallization using ethers such as ethylene glycol dimethyl ether. This eliminates the dependency on column chromatography entirely. The subsequent conversion of this high-purity solid into the final bromide target ensures that the final product inherits this exceptional purity profile, consistently achieving HPLC purity levels above 99%. For supply chain heads, this means a transition from a batch-process bottleneck to a streamlined, scalable workflow that significantly enhances throughput capabilities and reduces the environmental footprint associated with excessive solvent usage.
By converting the terminal hydroxyl group into a mesylate, the process creates a molecule with distinct lattice energy properties that allow for purification via simple solvent recrystallization using ethers such as ethylene glycol dimethyl ether. This eliminates the dependency on column chromatography entirely. The subsequent conversion of this high-purity solid into the final bromide target ensures that the final product inherits this exceptional purity profile, consistently achieving HPLC purity levels above 99%. For supply chain heads, this means a transition from a batch-process bottleneck to a streamlined, scalable workflow that significantly enhances throughput capabilities and reduces the environmental footprint associated with excessive solvent usage.
Mechanistic Insights into Sequential Nucleophilic Substitution and Sulfonate Activation
The chemical elegance of this synthesis lies in its controlled manipulation of nucleophilicity and leaving group ability across four distinct stages. The process begins with the deprotonation of 2,6-dichlorobenzyl alcohol using a strong base like sodium hydride in tetrahydrofuran, generating a reactive alkoxide species. This nucleophile attacks a dihaloethane (such as 1,2-dibromoethane or 1,2-diiodoethane) in an SN2 fashion to form the initial ether linkage (Formula IV). The choice of halogen in the dihaloalkane is critical; while bromine is preferred for cost, iodine offers enhanced reactivity, allowing the reaction to proceed efficiently at room temperature with yields approaching 90%. The second stage extends the carbon chain by reacting Formula IV with 1,6-hexanediol. Here, the remaining halogen on the ethyl chain acts as the electrophile, displaced by one of the hydroxyl groups of the diol under basic conditions (potassium tert-butoxide) at moderate temperatures (40-60°C). This step is meticulously controlled to prevent double substitution or polymerization, ensuring the terminal hydroxyl group remains available for the crucial activation step.
The third and most mechanistically significant step is the sulfonate esterification of the terminal alcohol (Formula III) to form the mesylate (Formula II). By reacting the alcohol with methanesulfonyl chloride in the presence of an acid scavenger like N,N-diisopropylethylamine at low temperatures (0-5°C), the poor leaving group (-OH) is converted into an excellent leaving group (-OMs). This transformation not only activates the molecule for the final halogen exchange but also alters its physical properties to enable crystallization. The final step involves a Finkelstein-type halogen exchange where the mesylate is displaced by a bromide ion from lithium bromide in acetonitrile. The use of lithium bromide is advantageous due to its high solubility in polar aprotic solvents, driving the equilibrium toward the formation of the target alkyl bromide (Formula I). This mechanistic sequence ensures that impurities generated in early steps are effectively scrubbed out during the recrystallization of Intermediate II, resulting in a final product with a remarkably clean impurity profile suitable for sensitive API applications.
How to Synthesize 2-[2-(6-bromohexyloxy)ethoxymethyl]-1,3-dichlorobenzene Efficiently
The execution of this synthesis requires precise control over reaction parameters to maximize the benefits of the crystallization strategy. The process is divided into five operational units: initial etherification, chain extension, mesylation, recrystallization, and final bromination. Each unit demands specific solvent systems and temperature profiles to ensure safety and yield optimization. For instance, the exothermic nature of the hydride deprotonation requires careful addition rates, while the mesylation step demands strict temperature control to prevent side reactions. The following guide summarizes the standardized operational flow derived from the patent examples, providing a roadmap for process engineers to implement this technology.
- Perform nucleophilic substitution of 2,6-dichlorobenzyl alcohol with dihaloethane using sodium hydride to form the initial ether linkage.
- React the resulting intermediate with 1,6-hexanediol under basic conditions to extend the carbon chain.
- Convert the terminal hydroxyl group to a mesylate solid intermediate, purify via recrystallization, and finally substitute with lithium bromide to yield the target bromide.
Commercial Advantages for Procurement and Supply Chain Teams
For stakeholders focused on the bottom line and supply resilience, the technical improvements in CN115286491A translate directly into tangible commercial benefits. The elimination of column chromatography is perhaps the most significant value driver. Chromatography is a major cost center in fine chemical manufacturing, consuming vast quantities of silica gel and organic solvents while requiring specialized equipment and extensive labor. By replacing this with a simple recrystallization step, the process drastically simplifies the manufacturing workflow. This simplification leads to substantial cost savings in manufacturing, as the capital expenditure for chromatography columns and the operational expenditure for solvent recovery are significantly reduced. Furthermore, the use of 2,6-dichlorobenzyl alcohol as a starting material offers a cost advantage over the corresponding bromide, as alcohols are generally more stable, easier to handle, and commercially available at lower price points from bulk chemical suppliers.
- Cost Reduction in Manufacturing: The shift from chromatographic purification to crystallization fundamentally alters the cost structure of producing this intermediate. Crystallization is a unit operation that scales linearly and efficiently, whereas chromatography scales poorly and is resource-intensive. By removing the need for silica gel and reducing solvent volumes by orders of magnitude, the variable cost per kilogram of product is significantly lowered. Additionally, the high purity achieved (>99%) reduces the need for re-processing or blending, further enhancing yield efficiency. The avoidance of expensive transition metal catalysts or exotic reagents in favor of commodity chemicals like methanesulfonyl chloride and lithium bromide ensures that raw material costs remain stable and predictable, shielding the supply chain from volatility in specialty chemical markets.
- Enhanced Supply Chain Reliability: The ability to produce a solid intermediate (Formula II) introduces a strategic buffer into the supply chain. Solids are inherently more stable and easier to store and transport than oils, reducing the risk of degradation during logistics. This stability allows manufacturers to stockpile key intermediates, decoupling production schedules from immediate demand fluctuations and ensuring continuous availability for downstream API synthesis. Moreover, the robustness of the reaction conditions—operating at mild temperatures and using common solvents like THF, acetonitrile, and ethers—means that the process can be easily transferred between different manufacturing sites without requiring specialized infrastructure. This flexibility mitigates the risk of single-source bottlenecks and enhances the overall resilience of the pharmaceutical supply network.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this route offers a greener alternative to traditional methods. The reduction in solvent usage directly correlates with a lower carbon footprint and reduced waste disposal costs. Crystallization generates less hazardous waste compared to silica-laden chromatography fractions, simplifying waste treatment protocols. The process is highly amenable to scale-up, moving seamlessly from kilogram to multi-ton production without the engineering challenges associated with large-scale chromatography. This scalability ensures that the method can meet the growing global demand for Vilanterol-based therapies without compromising on quality or compliance with increasingly stringent environmental regulations regarding solvent emissions and chemical waste.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. These insights are derived directly from the experimental data and claims within patent CN115286491A, providing clarity on the feasibility and advantages of the technology. Understanding these nuances is essential for technical teams evaluating the adoption of this process for commercial production.
Q: Why is the new synthesis route for Vilanterol intermediate considered superior to conventional methods?
A: The new route utilizes 2,6-dichlorobenzyl alcohol instead of the unstable benzyl bromide, generating a solid mesylate intermediate that allows for purification via recrystallization rather than difficult column chromatography, significantly improving purity and scalability.
Q: What are the critical purification advantages of Intermediate II in this process?
A: Intermediate II is a solid compound that can be refined using ether solvents like ethylene glycol dimethyl ether, achieving purity levels exceeding 99% without the need for silica gel column chromatography, which is a major bottleneck in industrial production.
Q: How does this method impact the cost structure of respiratory API manufacturing?
A: By eliminating the need for expensive chromatographic purification and utilizing cheaper, more stable raw materials like benzyl alcohol, the process drastically reduces solvent consumption and operational complexity, leading to substantial cost savings in large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-[2-(6-bromohexyloxy)ethoxymethyl]-1,3-dichlorobenzene Supplier
The technological advancements detailed in CN115286491A underscore the critical importance of selecting a manufacturing partner with deep expertise in process optimization and scale-up. NINGBO INNO PHARMCHEM stands at the forefront of this capability, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our commitment to quality is unwavering, supported by stringent purity specifications and rigorous QC labs that ensure every batch of 2-[2-(6-bromohexyloxy)ethoxymethyl]-1,3-dichlorobenzene meets the highest industry standards. We understand that the transition from a laboratory patent to a commercial reality requires more than just chemical knowledge; it demands engineering precision and a proactive approach to supply chain management. Our team is equipped to handle the complexities of crystallization-based processes, ensuring that the theoretical benefits of this new route are fully realized in practical, large-scale operations.
We invite global pharmaceutical partners to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. By leveraging this optimized synthesis route, we can help you achieve significant reductions in COGS while securing a stable supply of high-purity intermediates. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments. Together, we can accelerate the development of next-generation respiratory therapies, ensuring that life-saving medications reach patients faster and more affordably. Let us be your strategic partner in navigating the complexities of modern API manufacturing.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →