Advanced Five-Step Synthesis Strategy For High-Purity Vitamin D Drug Intermediates
Introduction To Patented Synthesis Technology
The pharmaceutical industry continuously seeks more efficient pathways for synthesizing complex active pharmaceutical ingredients, particularly for high-value therapeutics like Vitamin D analogs. Patent CN106916181B, published in October 2020, introduces a groundbreaking preparation method for key intermediates of Vitamin D drugs, specifically targeting the synthesis of the compound designated as Formula XI. This technology addresses critical bottlenecks in the manufacturing of Calcipotriol and its derivatives, which are essential for treating psoriasis and other dermatological conditions. By re-engineering the synthetic route starting from Inhoffen-Lythgoe diol, the inventors have successfully condensed an traditionally lengthy eight-step process into a highly efficient five-step sequence. This reduction in step count is not merely a numerical improvement but represents a fundamental shift in process chemistry that enhances overall yield, reduces solvent consumption, and simplifies purification protocols. For R&D directors and procurement specialists, understanding the nuances of this patent is vital for securing a reliable supply chain of high-purity intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of the cyclopropylenone side chain and its connection to the CD and A rings of Vitamin D analogs has been fraught with chemical challenges. Traditional routes, such as those described in earlier literature like WO 87/00834, often involve connecting a precursor like Formula I with a side chain like Formula IIa or IIb. A major drawback of these legacy methods is the necessity for chiral reduction after the side chain connection, where the presence of polyene structures negatively impacts stereoselectivity and yield. Furthermore, these routes frequently require photoisomerization to invert the 5,6-double bond, a step that severely limits product isolation efficiency and batch scale-up capabilities. Even alternative approaches attempting to couple Formula III with Formula IV have struggled with the synthesis of Fragment III itself.
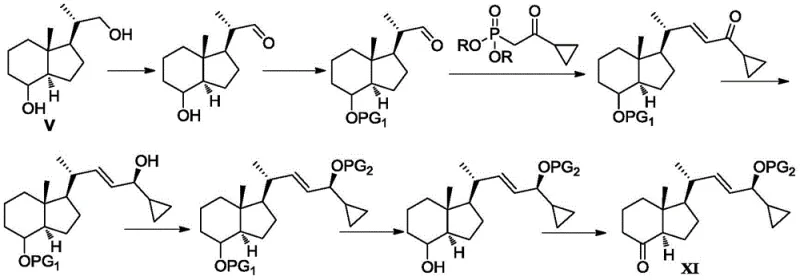
As illustrated in the conventional pathway, the synthesis of Fragment III from Inhoffen-Lythgoe diol (Formula V) typically demands eight distinct reaction steps. This arduous journey involves selective oxidation of primary alcohols, followed by hydroxyl protection, Horner-Wadsworth-Emmons reactions, chiral reduction, protection of chiral hydroxyls, selective deprotection, and final oxidation. The reliance on multiple protection and deprotection cycles introduces significant opportunities for yield loss and impurity generation. Moreover, the frequent requirement for column chromatography purification at various stages makes the process time-consuming, expensive, and notoriously difficult to translate from laboratory benchtop to industrial reactor scales.
The Novel Approach
In stark contrast, the novel methodology disclosed in CN106916181B offers a streamlined solution that bypasses these historical inefficiencies. The core innovation lies in the strategic selection of reagents and protecting groups that allow for direct transformation without unnecessary intermediate manipulations. By utilizing a triarylphosphine ylide (Formula VII) instead of traditional phosphonate esters, the reaction with the aldehyde (Formula VI) proceeds with high selectivity, avoiding unwanted side reactions with the ring ketone carbonyl. This clever chemical design eliminates the need to protect the ring hydroxyl group prior to the Wittig coupling. Additionally, the use of sterically bulky hydroxyl protecting agents enables selective protection of the specific hydroxyl group on the asterisk carbon in Formula IX, thereby removing the need for complex selective deprotection steps later in the sequence.
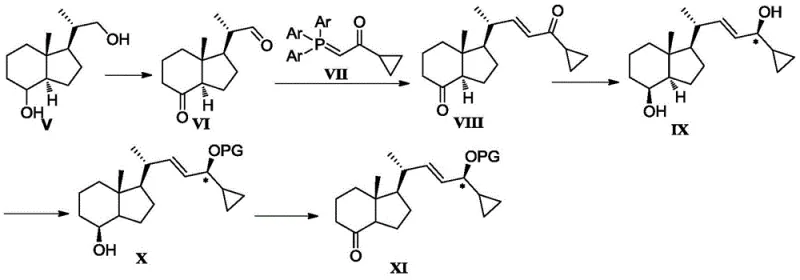
The result is a robust five-step synthesis that converts Formula V directly to Formula XI. This route is characterized by its operational simplicity and high yield, achieving satisfactory results through crystallization separation rather than labor-intensive column chromatography. The elimination of redundant steps not only accelerates the production timeline but also drastically reduces the consumption of raw materials and solvents. For a reliable vitamin D intermediate supplier, adopting this methodology translates to a more stable and cost-effective manufacturing process, ensuring consistent quality and availability for downstream pharmaceutical applications.
Mechanistic Insights Into The Streamlined Synthesis Route
The success of this five-step synthesis relies heavily on precise control over reaction conditions and reagent stoichiometry. The process initiates with the oxidation of Inhoffen-Lythgoe diol (Formula V) to the corresponding aldehyde (Formula VI). This step is carefully managed using oxidants such as Pyridinium Chlorochromate (PCC) or Jones reagent in solvents like dichloromethane, often buffered with basic compounds like sodium acetate to maintain optimal pH levels. Following this, the critical Wittig reaction couples Formula VI with the triarylphosphine ylide Formula VII. Unlike phosphonates which might react indiscriminately, the triarylphosphine reagent demonstrates remarkable chemoselectivity for the aldehyde moiety even in the presence of other reactive functional groups, yielding the enone Formula VIII with high purity.
Subsequent steps focus on establishing the correct stereochemistry and functionality required for the final drug molecule. The chiral reduction of Formula VIII to Formula IX is performed using a CBS (Corey-Bakshi-Shibata) catalyst system, typically employing (S)-2-methyl-CBS-oxazaborane and a borane reducing agent at controlled low temperatures ranging from -40°C to 25°C. This ensures the formation of the desired R-configuration at the asterisk carbon. The final stages involve selective protection of this newly formed hydroxyl group using bulky silyl agents like tert-butyldimethylsilyl chloride, followed by a final oxidation to generate the ketone in Formula XI. Throughout this sequence, the avoidance of column chromatography is achieved by optimizing crystallization conditions, allowing impurities to remain in the mother liquor while the product precipitates in high purity.
How to Synthesize Formula XI Compound Efficiently
Implementing this synthesis protocol requires strict adherence to the optimized reaction parameters outlined in the patent to ensure reproducibility and high yield. The process is designed to be scalable, moving seamlessly from gram-scale laboratory experiments to multi-kilogram production batches. Operators must pay close attention to temperature controls during the chiral reduction phase and the stoichiometric ratios during the oxidation steps to minimize byproduct formation. The detailed standard operating procedures for each of the five steps, including specific solvent choices, reaction times, and workup protocols, are essential for maintaining the integrity of the chiral centers and the overall purity of the intermediate.
- Oxidize Inhoffen-Lythgoe diol (Formula V) using PCC or Jones reagent to obtain aldehyde Formula VI.
- Perform Wittig reaction between Formula VI and triarylphosphine ylide Formula VII to generate enone Formula VIII.
- Execute chiral reduction of Formula VIII using CBS catalyst and borane to yield chiral alcohol Formula IX.
- Selectively protect the hydroxyl group on the asterisk carbon of Formula IX using a bulky silyl protecting agent to form Formula X.
- Oxidize Formula X using PDC or similar oxidants to obtain the final ketone intermediate Formula XI.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition from an eight-step legacy process to this novel five-step route offers substantial strategic benefits beyond mere technical elegance. The primary advantage lies in the significant reduction of manufacturing costs driven by the decreased number of unit operations. Fewer reaction steps inherently mean less labor, reduced energy consumption for heating and cooling, and lower solvent usage for both reaction and purification. Furthermore, the elimination of column chromatography is a game-changer for cost reduction in pharmaceutical intermediate manufacturing, as chromatographic purification is often the most expensive and time-consuming part of fine chemical production, requiring vast amounts of silica gel and solvents.
- Cost Reduction in Manufacturing: The streamlined five-step process drastically cuts down on raw material consumption and waste generation. By avoiding multiple protection and deprotection cycles, the process reduces the quantity of reagents such as silyl chlorides and deprotecting acids or fluorides needed. The ability to purify intermediates via crystallization rather than chromatography significantly lowers the cost of goods sold (COGS), making the final Vitamin D intermediate more price-competitive in the global market. This efficiency allows suppliers to offer better pricing structures without compromising on margin, providing a clear economic advantage to downstream drug manufacturers.
- Enhanced Supply Chain Reliability: Simplifying the synthesis route directly correlates to improved supply chain resilience. With fewer steps, there are fewer points of failure where a reaction could stall or yield poor quality material, thereby reducing the risk of batch failures. The use of common, commercially available reagents like PCC, PDC, and standard silyl protecting groups ensures that raw material sourcing is stable and not dependent on exotic or hard-to-find chemicals. This reliability shortens the lead time for high-purity pharmaceutical intermediates, allowing procurement teams to plan inventory more effectively and respond faster to market demand fluctuations.
- Scalability and Environmental Compliance: The design of this process is inherently suited for commercial scale-up of complex pharmaceutical intermediates. The avoidance of column chromatography not only saves money but also aligns with green chemistry principles by minimizing solvent waste, which is a critical factor for environmental compliance in modern chemical manufacturing. The robust nature of the reactions, which tolerate standard industrial conditions, facilitates the transfer from pilot plants to large-scale production facilities. This scalability ensures that suppliers can meet increasing volume requirements for Vitamin D drugs as global demand for psoriasis treatments continues to grow.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of Vitamin D intermediates using the methodology described in patent CN106916181B. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the feasibility and advantages of this specific chemical route. Understanding these details helps stakeholders make informed decisions regarding technology adoption and supplier selection.
Q: How does this new synthesis route improve upon conventional methods for Vitamin D intermediates?
A: The conventional route typically requires eight steps involving multiple protection and deprotection sequences, often necessitating column chromatography. This patented method reduces the process to five steps by utilizing a triarylphosphine reagent that selectively reacts with the aldehyde group without prior ring hydroxyl protection, and employs bulky protecting groups for selective mono-protection, eliminating complex purification steps.
Q: What specific reagents are used to ensure high stereoselectivity in the reduction step?
A: The process utilizes a chiral reduction strategy involving a CBS catalyst, specifically (S)-2-methyl-CBS-oxazaborane or (S)-2-isopropyl-CBS-oxazaborane, combined with a borane reducing agent such as borane dimethyl sulfide. This system ensures high stereocontrol at the asterisk carbon, crucial for the biological activity of the final Vitamin D analog.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the method is explicitly designed for industrial scalability. It avoids column chromatography entirely, relying instead on crystallization and simple filtration for purification. The use of robust reagents like PCC and PDC, along with simplified operational steps, significantly lowers production costs and facilitates batch scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Calcipotriol Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthesis routes in the competitive landscape of pharmaceutical intermediates. Our technical team has thoroughly analyzed the innovations presented in CN106916181B and possesses the expertise to implement this advanced five-step synthesis for the commercial production of Formula XI. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are realized in practical, large-volume manufacturing. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of verifying the stereochemical integrity and chemical purity of every batch, guaranteeing that our clients receive intermediates that meet the highest global regulatory standards.
We invite pharmaceutical companies and research institutions to collaborate with us to leverage this cost-effective and scalable technology. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific production volumes and quality requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you optimize your supply chain for Vitamin D drug manufacturing with a partner dedicated to quality, efficiency, and long-term reliability.