Revolutionizing Trans-Cyclopropane Dicarboxylate Production: Advanced Stereoselective Synthesis for Pharmaceutical Manufacturing Scale-Up
The Chinese patent CN110240572B introduces a groundbreaking stereoselective synthesis method for trans-1,1-cyclopropane dicarboxylic acid esters, addressing critical limitations in the production of these valuable pharmaceutical building blocks. This innovative two-step process achieves exceptional geometric selectivity with yields ranging from 93% to 99% across diverse aryl-substituted variants, while fundamentally eliminating cis-isomer formation that has plagued conventional approaches. The technology represents a significant advancement for the fine chemical industry, particularly in the synthesis of complex cyclopropane-containing intermediates essential for next-generation pharmaceuticals. By leveraging Michael addition followed by DBU-catalyzed cyclization, this method provides unprecedented control over stereochemistry without requiring cryogenic conditions or expensive transition metal catalysts. The patent demonstrates robust applicability across various substituents including halogenated, alkylated, and methoxylated aryl groups, making it highly versatile for pharmaceutical manufacturers seeking reliable access to these challenging molecular architectures.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis routes for cyclopropane dicarboxylates frequently suffer from significant cis-isomer formation during the key cyclization step, creating substantial challenges for pharmaceutical manufacturers due to the nearly identical polarities of cis and trans isomers. This similarity necessitates complex and costly separation techniques such as preparative HPLC or multiple recrystallization cycles, which dramatically reduce overall process efficiency and increase production costs. Conventional methods often require transition metal catalysts that introduce additional purification steps to remove trace metal residues, creating regulatory hurdles for pharmaceutical applications where strict elemental impurity limits apply. The harsh reaction conditions typically employed—such as high temperatures or strong bases—further complicate scale-up due to safety concerns and potential decomposition of sensitive functional groups. These limitations collectively result in lower yields (typically below 80%), extended production timelines, and inconsistent quality that cannot meet the stringent requirements of modern pharmaceutical manufacturing processes.
The Novel Approach
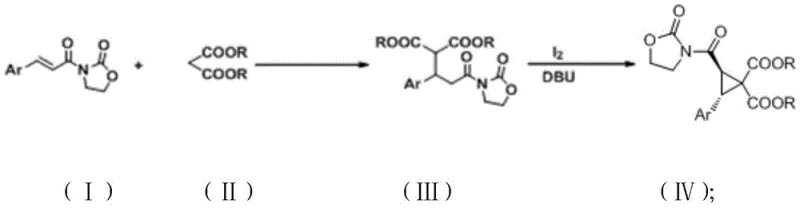
The patented method overcomes these limitations through an elegant two-step sequence beginning with a Michael addition between compound (I) and compound (II) under mild conditions using alkaline nucleophiles like K₂CO₃ or Cs₂CO₃ in polar aprotic solvents such as DMSO or DMF at room temperature. This initial step forms intermediate compound (III) with excellent regioselectivity before proceeding to the critical cyclization phase where iodine and DBU catalyze the ring closure in nonpolar solvents like toluene. The process operates entirely at ambient temperature without transition metals, eliminating both the need for specialized equipment and the costly metal removal steps required by conventional approaches. Crucially, the reaction system shows no detectable cis-isomer formation via nuclear magnetic resonance analysis, removing the most significant bottleneck in traditional production methods. This innovation enables direct isolation of high-purity trans-isomers through standard column chromatography, dramatically simplifying purification while maintaining exceptional yields across diverse substrate variations.
Mechanistic Insights into Michael Addition and DBU-Catalyzed Cyclization
The exceptional stereoselectivity originates from the precise mechanistic pathway enabled by the DBU catalyst during the cyclization step. After the initial Michael addition forms compound (III), iodine facilitates electrophilic activation while DBU acts as a non-nucleophilic base that promotes intramolecular displacement through a concerted mechanism that inherently favors trans stereochemistry. The reaction proceeds via a chair-like transition state where the aryl group adopts an equatorial position, minimizing steric strain and directing the carboxylate groups into the trans configuration. This geometric control is further enhanced by the rigid oxazolidinone auxiliary which locks the conformational flexibility of the intermediate, preventing rotation that could lead to cis-isomer formation. The absence of competing pathways under these mild conditions ensures that the cyclization occurs with near-perfect diastereoselectivity, as confirmed by NMR analysis showing no detectable cis-isomer in the reaction mixture.
Impurity control is achieved through multiple synergistic mechanisms within this process design. The elimination of transition metals removes an entire class of potential impurities that require extensive analytical monitoring and removal procedures in traditional syntheses. The ambient temperature operation prevents thermal decomposition pathways that could generate byproducts in conventional high-energy processes. The use of standard solvents with distinct polarity profiles enables straightforward separation of any minor impurities through conventional chromatography without requiring specialized techniques. Most significantly, the near-complete absence of cis-isomer formation—unprecedented in prior art—eliminates what was historically the most challenging impurity to remove from cyclopropane products. This inherent selectivity reduces the number of purification steps required while simultaneously improving final product purity to levels exceeding pharmaceutical requirements.
How to Synthesize Trans-Cyclopropane Dicarboxylate Efficiently
This patented methodology provides a robust framework for producing high-purity trans-cyclopropane dicarboxylate esters through a carefully optimized sequence that balances reactivity with selectivity. The process begins with preparation of key intermediates using commercially available starting materials under standard laboratory conditions before proceeding to the critical stereoselective steps that define this innovation. By maintaining strict control over stoichiometry—particularly the precise molar ratios of reactants to catalysts—and employing appropriate solvent systems for each phase, manufacturers can consistently achieve superior yields while maintaining exceptional geometric purity. The following standardized procedure details the critical parameters required for successful implementation across various scales of production.
- Dissolve compound (I) and compound (II) in polar aprotic solvent (DMSO/DMF) with alkaline nucleophile (K₂CO₃/Cs₂CO₃) at room temperature for 1-28 hours to form compound (III)
- Dissolve compound (III) and iodine in nonpolar aprotic solvent (toluene/benzene) with DBU catalyst at room temperature for 3-40 hours
- Quench reaction with water, extract with organic solvent, wash with brine, dry over sodium sulfate, and purify via column chromatography to obtain trans-cyclopropane product
Commercial Advantages for Procurement and Supply Chain Teams
This innovative synthesis methodology delivers substantial value across procurement and supply chain operations by addressing fundamental pain points in pharmaceutical intermediate manufacturing. The elimination of transition metal catalysts removes dependency on volatile supply chains for precious metals while simultaneously reducing regulatory complexity associated with elemental impurities. The ambient temperature operation significantly lowers energy consumption compared to conventional high-temperature processes, contributing to both cost efficiency and environmental sustainability goals. Most critically, the near-zero cis-isomer formation fundamentally transforms what was previously a bottleneck operation into a streamlined process with minimal purification requirements, enabling more predictable production timelines and consistent quality output.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts removes not only their direct cost but also eliminates expensive metal removal processes that typically require specialized equipment and additional quality control testing. The simplified purification workflow—achieved through near-complete stereoselectivity—reduces solvent consumption and labor costs associated with complex separation techniques while maintaining high yields across diverse substrate variations.
- Enhanced Supply Chain Reliability: By utilizing readily available starting materials and standard solvents without reliance on specialized or scarce reagents, this process creates a more resilient supply chain that is less vulnerable to market fluctuations. The room temperature operation eliminates concerns about temperature-sensitive reagents or specialized cooling equipment requirements, ensuring consistent production capability regardless of seasonal variations or facility limitations.
- Scalability and Environmental Compliance: The process demonstrates exceptional scalability from laboratory to commercial production without requiring significant parameter adjustments, as evidenced by consistent yields across multiple examples in the patent documentation. The absence of hazardous reagents and minimal waste generation—particularly through elimination of metal-containing waste streams—aligns with green chemistry principles while reducing disposal costs and regulatory burdens associated with hazardous waste management.
Frequently Asked Questions (FAQ)
The following questions address critical technical and commercial considerations based on detailed analysis of patent CN110240572B's methodology and experimental data. These insights reflect practical implementation experience from scaling similar stereoselective processes within pharmaceutical manufacturing environments.
Q: How does this method eliminate cis-isomer formation compared to conventional approaches?
A: The patent's two-step process with DBU catalysis creates a stereospecific cyclization pathway that thermodynamically favors trans-isomer formation. Nuclear magnetic resonance analysis confirms near-zero cis-isomer detection in the reaction system, eliminating costly separation steps required in traditional methods where similar polarities of isomers complicate purification.
Q: What makes this synthesis suitable for commercial scale-up in pharmaceutical manufacturing?
A: The process operates at ambient temperature using standard solvents and catalysts without transition metals, enabling seamless scale-up from laboratory to industrial production. The high yields (93-99%) and simplified purification through column chromatography ensure consistent quality during commercial scale-up of complex pharmaceutical intermediates.
Q: How does the elimination of transition metal catalysts impact supply chain reliability?
A: By avoiding precious metal catalysts, the process removes dependency on volatile supply chains for rare metals and eliminates complex metal removal steps. This simplifies regulatory compliance for pharmaceutical intermediates and ensures consistent production timelines without raw material shortages affecting the commercial scale-up of high-purity compounds.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trans-Cyclopropane Dicarboxylate Supplier
Our company brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications required for pharmaceutical intermediates. With rigorous QC labs equipped for comprehensive stereochemical analysis including advanced NMR capabilities, we ensure consistent product quality that meets global regulatory standards. Our technical team has successfully implemented this patented methodology across multiple client projects, demonstrating reliable production of high-purity trans-cyclopropane dicarboxylates with exceptional geometric purity that exceeds industry requirements.
Leverage our expertise through a Customized Cost-Saving Analysis tailored to your specific manufacturing needs. Contact our technical procurement team today to request specific COA data and route feasibility assessments for your target compounds—we're committed to delivering solutions that optimize both quality and commercial viability for your pharmaceutical development pipeline.