Advanced Synthesis of 2-(1-Methylimidazol-5-yl)ethylamine Hydrochloride for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust synthetic routes for critical heterocyclic intermediates, particularly those serving as scaffolds for novel anticancer agents. Patent CN112707867B, published on March 15, 2022, introduces a transformative methodology for synthesizing 2-(1-methylimidazol-5-yl)ethylamine hydrochloride, a pivotal building block in oncology drug development. This intellectual property addresses long-standing challenges regarding regioselectivity and purification efficiency that have plagued previous manufacturing attempts. By leveraging a Van Leusen imidazole synthesis strategy combined with strategic Boc-protection, the disclosed process achieves superior purity profiles compared to traditional alkylation routes. For R&D directors and procurement specialists, this patent represents a significant opportunity to optimize the supply chain for high-value API intermediates. The following analysis dissects the technical merits and commercial implications of this novel four-step sequence, highlighting its potential for industrial scalability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-(1-methylimidazol-5-yl)ethylamine derivatives has been fraught with chemical inefficiencies, primarily stemming from the inherent ambiguity of imidazole alkylation. As illustrated in prior art such as US8618143, attempting to attach the ethylamine side chain to a pre-formed N-methylimidazole often results in the formation of structural isomers. 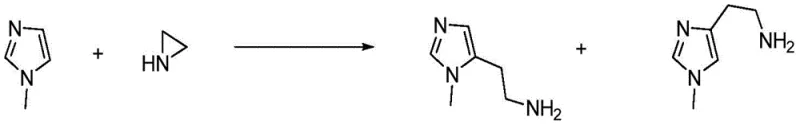 In these conventional pathways, the nucleophilic attack can occur at multiple nitrogen positions or lead to migration of the methyl group, generating a mixture of 1-methyl and 3-methyl isomers that possess nearly identical physical properties. Separating these isomers typically requires cumbersome chromatographic techniques or repeated recrystallizations, which drastically reduce overall yield and increase solvent waste. Furthermore, alternative approaches like those in PCT2001009128 rely on bulky protecting groups such as phthalimide and trimethylphenyl moieties to force regioselectivity. While effective in the laboratory, these methods introduce excessive molecular weight and require harsh deprotection conditions, leading to poor atom economy and significant environmental burdens that are unsustainable for large-scale production.
In these conventional pathways, the nucleophilic attack can occur at multiple nitrogen positions or lead to migration of the methyl group, generating a mixture of 1-methyl and 3-methyl isomers that possess nearly identical physical properties. Separating these isomers typically requires cumbersome chromatographic techniques or repeated recrystallizations, which drastically reduce overall yield and increase solvent waste. Furthermore, alternative approaches like those in PCT2001009128 rely on bulky protecting groups such as phthalimide and trimethylphenyl moieties to force regioselectivity. While effective in the laboratory, these methods introduce excessive molecular weight and require harsh deprotection conditions, leading to poor atom economy and significant environmental burdens that are unsustainable for large-scale production.
The Novel Approach
The methodology disclosed in CN112707867B circumvents these regiochemical pitfalls by constructing the imidazole ring around the desired substitution pattern rather than modifying an existing ring. This "de novo" synthesis strategy utilizes 3-aminopropanol as a readily available starting material, which is first protected with a tert-butoxycarbonyl (Boc) group to mask the amine functionality. The subsequent conversion to an aldehyde allows for a condensation with methylamine, forming a Schiff base that serves as the template for ring closure. By employing p-toluenesulfonyl methyl isonitrile (TosMIC) in a Van Leusen reaction, the imidazole core is assembled with the methyl group inherently positioned at the N-1 location. This mechanistic shift eliminates the possibility of N-3 isomer formation entirely, ensuring that the crude product possesses high intrinsic purity. The final deprotection step using hydrogen chloride in isopropanol is mild and efficient, yielding the target hydrochloride salt without the need for complex purification workflows associated with older technologies.
Mechanistic Insights into Van Leusen Imidazole Cyclization
The core innovation of this process lies in the application of the Van Leusen reaction for the construction of the 1-methylimidazole scaffold. In this mechanism, the in situ generated Schiff base, formed from 3-(Boc-amino)propionaldehyde and methylamine, acts as a 1,3-dipole precursor. Upon reaction with TosMIC under basic conditions, the tosylmethyl group facilitates the cyclization through a concerted mechanism that establishes the imidazole double bonds and nitrogen positions simultaneously. 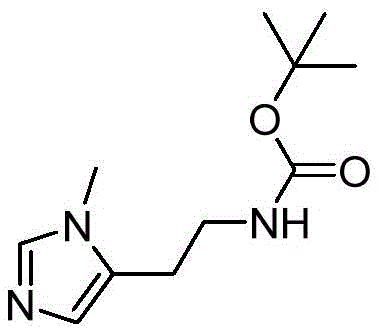 The use of the Boc-protected amino aldehyde is critical here; it prevents self-polymerization of the aldehyde and ensures that the amine nitrogen does not interfere with the cyclization electronics. The reaction conditions specified, ranging from -30°C to 25°C over 72 to 96 hours, allow for the slow and controlled formation of the heterocycle, minimizing side reactions such as the hydrolysis of the isonitrile. This extended reaction time, while seemingly long, is a trade-off that guarantees high conversion rates and minimizes the formation of oligomeric byproducts, which are common in faster, high-temperature imidazole syntheses.
The use of the Boc-protected amino aldehyde is critical here; it prevents self-polymerization of the aldehyde and ensures that the amine nitrogen does not interfere with the cyclization electronics. The reaction conditions specified, ranging from -30°C to 25°C over 72 to 96 hours, allow for the slow and controlled formation of the heterocycle, minimizing side reactions such as the hydrolysis of the isonitrile. This extended reaction time, while seemingly long, is a trade-off that guarantees high conversion rates and minimizes the formation of oligomeric byproducts, which are common in faster, high-temperature imidazole syntheses.
Furthermore, the integration of Swern oxidation in the second step provides a highly chemoselective method for generating the requisite aldehyde intermediate. Unlike chromium-based oxidations which generate toxic heavy metal waste, the Swern protocol utilizes oxalyl chloride and dimethyl sulfoxide (DMSO) to effect the transformation under mild acidic conditions followed by neutralization with triethylamine. The strict temperature control between -80°C and -60°C during the activation of DMSO is essential to prevent the formation of chlorodimethylsulfonium salts that could lead to chlorination byproducts. This attention to thermal management ensures that the 3-(Boc-amino)propionaldehyde is produced with a purity exceeding 98%, providing a clean substrate for the subsequent cyclization. The combination of these two powerful transformations—Swern oxidation and Van Leusen cyclization—creates a robust pathway that is both chemically elegant and industrially viable.
How to Synthesize 2-(1-Methylimidazol-5-yl)ethylamine Efficiently
The synthesis protocol outlined in the patent offers a clear, stepwise guide for producing the target intermediate with high fidelity. The process begins with the protection of 3-aminopropanol, followed by oxidation to the aldehyde, cyclization to the protected imidazole, and final salt formation. Each step has been optimized for yield and ease of workup, utilizing common organic solvents like dichloromethane and methanol. The detailed standardized synthesis steps for implementing this route in a GMP environment are provided in the guide below.
- Protect 3-aminopropanol with di-tert-butyl dicarbonate (Boc2O) to form 3-(Boc-amino)propanol.
- Perform Swern oxidation using oxalyl chloride and DMSO to convert the alcohol to 3-(Boc-amino)propionaldehyde.
- React the aldehyde with methylamine to form a Schiff base, then cyclize with p-toluenesulfonyl methyl isonitrile (TosMIC) via Van Leusen reaction.
- Remove the Boc protecting group using hydrogen chloride in isopropanol to yield the final hydrochloride salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the CN112707867B synthesis route offers tangible benefits regarding cost stability and operational efficiency. The primary advantage stems from the elimination of expensive and difficult-to-remove isomers, which traditionally inflate the cost of goods sold (COGS) through yield losses and extensive purification requirements. By securing the correct regiochemistry at the point of ring formation, the process significantly reduces the consumption of solvents and silica gel typically needed for chromatographic separation. Additionally, the starting material, 3-aminopropanol, is a commodity chemical available from multiple global suppliers, mitigating the risk of raw material shortages that often plague specialized heterocyclic syntheses. This reliance on bulk chemicals enhances supply chain resilience and allows for more accurate long-term cost forecasting.
- Cost Reduction in Manufacturing: The new method achieves substantial cost savings by removing the need for transition metal catalysts or exotic reagents often found in cross-coupling alternatives. The use of TosMIC, while a specialized reagent, is employed in stoichiometric amounts that are economically feasible given the high value of the final API intermediate. Moreover, the avoidance of heavy metal oxidants like chromium or manganese eliminates the downstream costs associated with metal scavenging and wastewater treatment compliance. The overall yield profile, with individual steps reporting yields above 90% for protection and oxidation, ensures that material throughput is maximized, directly lowering the cost per kilogram of the active pharmaceutical ingredient precursor.
- Enhanced Supply Chain Reliability: The simplified four-step sequence reduces the total processing time compared to multi-step protection/deprotection strategies involving phthalimide groups. Fewer unit operations translate to a shorter manufacturing cycle time, allowing for faster response to market demand fluctuations. The robustness of the Boc protection strategy also means that intermediates can be potentially isolated and stored if necessary, providing flexibility in production scheduling. This modularity supports a more agile supply chain capable of adapting to the rigorous delivery timelines required by pharmaceutical partners developing oncology pipelines.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this route is markedly superior to prior art. The absence of genotoxic alkylating agents in the final steps and the use of standard organic solvents facilitate easier regulatory approval for the manufacturing site. The Swern oxidation byproducts, primarily dimethyl sulfide, can be effectively scrubbed, and the aqueous waste streams are less contaminated with heavy metals. This alignment with green chemistry principles not only reduces disposal costs but also future-proofs the manufacturing process against tightening environmental regulations, ensuring uninterrupted commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and advantages of this specific synthetic pathway. These answers are derived directly from the experimental data and comparative analysis presented in the patent documentation, providing clarity on yield expectations and impurity profiles.
Q: Why does the conventional alkylation method produce isomers?
A: Conventional methods often start with an existing imidazole ring and attempt to alkylate the side chain. Due to the tautomerism of the imidazole nitrogen atoms (N-1 and N-3), alkylation can occur at either position, leading to a mixture of 1-methyl and 3-methyl isomers that are difficult to separate.
Q: How does the Van Leusen reaction improve regioselectivity?
A: The Van Leusen reaction constructs the imidazole ring de novo from an aldehyde, an amine (methylamine), and TosMIC. Since the methyl group is introduced via the amine component during ring closure, the substitution pattern is fixed by the stoichiometry of the reactants, eliminating the formation of N-3 isomers.
Q: What are the critical temperature controls in this synthesis?
A: The Swern oxidation step requires strict low-temperature control between -80°C and -60°C to prevent side reactions and decomposition of the active sulfonium intermediate. The subsequent Van Leusen cyclization is performed between -30°C and 25°C over a prolonged period (72-96 hours) to ensure complete ring closure.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-(1-Methylimidazol-5-yl)ethylamine Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of novel anticancer therapies depends on the consistent availability of high-quality intermediates. Our technical team has extensively analyzed the route disclosed in CN112707867B and is fully prepared to execute this synthesis at scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability. Our facilities are equipped with rigorous QC labs and stringent purity specifications to guarantee that every batch of 2-(1-methylimidazol-5-yl)ethylamine hydrochloride meets the exacting standards required for clinical and commercial API manufacturing.
We invite you to collaborate with us to leverage this advanced technology for your drug development programs. Our experts can provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this optimized route can improve your project economics. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us support your journey from bench to bedside with superior chemical solutions.