Advanced Synthesis of Anidulafungin Impurity F for Robust Quality Control and Commercial Scalability
The pharmaceutical landscape for echinocandin antifungals demands rigorous quality control, particularly for semi-synthetic agents like Anidulafungin (CAS: 166663-25-8). Patent CN112390723A introduces a pivotal advancement in the preparation of Anidulafungin Impurity Compound F, a critical degradation product that can compromise drug safety if not strictly monitored. This patent outlines a novel, high-yield synthetic route that transforms the production of this reference standard from a difficult isolation challenge into a straightforward, scalable chemical synthesis. By addressing the specific chemical origins of this impurity, the technology empowers manufacturers to establish robust analytical methods, ensuring that every batch of Anidulafungin released to the market meets the highest purity specifications required by global health authorities.
For R&D directors and quality assurance teams, the ability to access high-purity impurity standards is not merely a regulatory checkbox but a fundamental component of risk mitigation. The methodology described in CN112390723A leverages a deep understanding of the Anidulafungin side-chain chemistry to deliberately generate Impurity F with exceptional efficiency. This approach eliminates the variability associated with extracting trace impurities from complex fermentation broths or synthesis mixtures, providing a consistent and reliable supply of the reference material needed for method validation and stability testing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
In the traditional manufacturing of Anidulafungin, the formation of Impurity F is an unintended and often problematic side reaction. This impurity typically arises during the coupling of the echinocandin nucleus with the lipophilic side chain, specifically when industrial-grade triethylamine is employed as a base. Industrial triethylamine inevitably contains traces of monoethylamine and diethylamine as impurities due to the nature of its catalytic production from ethanol and ammonia. When these diethylamine contaminants encounter the activated ester intermediate of the Anidulafungin side chain, they compete with the intended nucleophile, leading to the formation of the diethylamide derivative known as Impurity F.
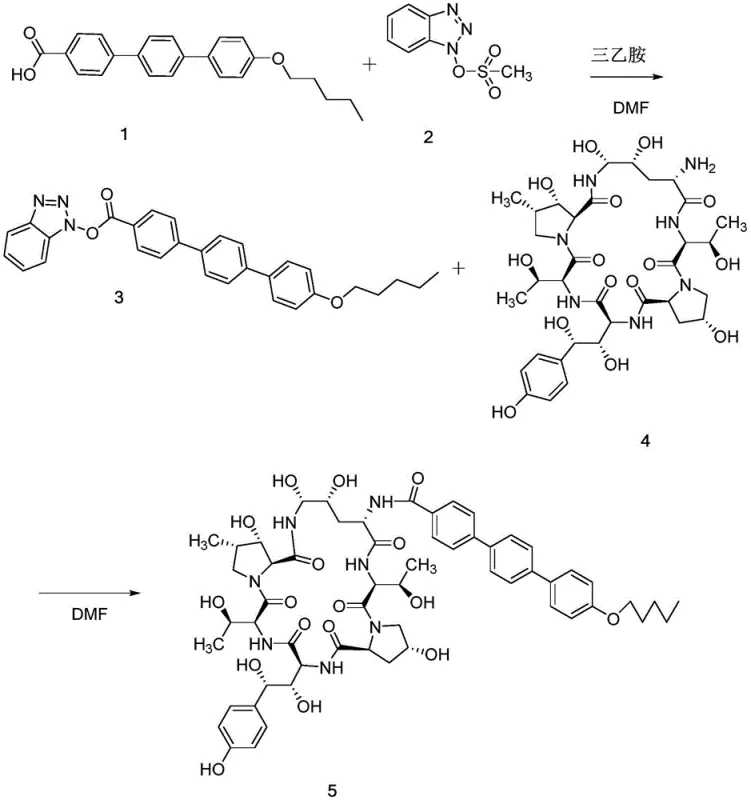
Historically, obtaining pure samples of Impurity F for analytical calibration has been a significant bottleneck. Chemists were forced to rely on isolating minute quantities of this byproduct from large-scale reaction mixtures, a process characterized by low recovery rates and tedious chromatographic purification. The resulting reference standards often suffered from inconsistent purity levels, making it difficult to establish accurate detection limits in HPLC assays. Furthermore, the reliance on accidental formation meant that supply was unpredictable; if a production batch had low levels of diethylamine contamination, there might be insufficient Impurity F generated to create a usable standard, thereby delaying quality control protocols and risking batch release timelines.
The Novel Approach
The innovation presented in patent CN112390723A fundamentally shifts the paradigm from accidental formation to intentional, controlled synthesis. Instead of hoping for the impurity to appear as a byproduct, the inventors have developed a dedicated pathway that reacts the Anidulafungin intermediate compound SM-I directly with pure diethylamine. This targeted approach bypasses the complexity of the full Anidulafungin synthesis, focusing solely on the transformation of the side-chain precursor. By controlling the stoichiometry and reaction conditions, the process ensures that the diethylamine reacts exclusively with the activated ester moiety of SM-I, driving the conversion to completion with minimal side reactions.
This deliberate synthesis strategy offers profound advantages in terms of yield and operational simplicity. The patent demonstrates that by selecting appropriate solvents such as dimethyl sulfoxide (DMSO), acetonitrile, or acetone, the reaction proceeds smoothly at mild temperatures ranging from 20°C to 30°C. The workup procedure is equally elegant, utilizing water precipitation to isolate the product, which avoids the need for expensive silica gel chromatography. This results in a final product with a molar yield consistently above 95% and a purity exceeding 98%, providing a superior reference standard that significantly enhances the reliability of impurity profiling in the final drug substance.
Mechanistic Insights into Nucleophilic Acyl Substitution
The chemical transformation at the heart of this patent is a classic nucleophilic acyl substitution, specifically the aminolysis of an activated ester. The starting material, SM-I, features a benzotriazole ester group attached to the terphenyl backbone. Benzotriazole esters are highly reactive electrophiles because the benzotriazole anion is an excellent leaving group, stabilized by resonance within its aromatic heterocyclic structure. When diethylamine is introduced into the reaction mixture, the lone pair of electrons on the nitrogen atom acts as a potent nucleophile, attacking the carbonyl carbon of the ester linkage.
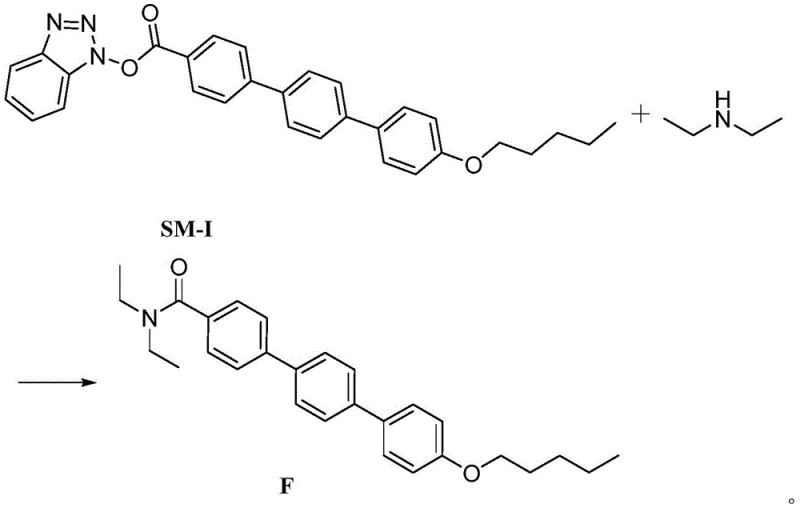
This nucleophilic attack leads to the formation of a tetrahedral intermediate, which subsequently collapses to expel the benzotriazole leaving group and form the stable amide bond found in Impurity F. The choice of solvent plays a critical role in facilitating this mechanism; polar aprotic solvents like DMSO or DMF stabilize the transition state and ensure that both the organic SM-I intermediate and the amine nucleophile remain in solution, maximizing collision frequency. The reaction is thermodynamically driven by the formation of the strong amide bond and the release of the stable benzotriazole byproduct. Understanding this mechanism allows process chemists to fine-tune reaction parameters, such as the molar ratio of diethylamine (typically 1:1 to 1:2 relative to SM-I), to ensure complete consumption of the starting material without generating excessive waste.
From an impurity control perspective, this mechanism highlights the specific vulnerability of the activated ester intermediate to secondary amines. In the main Anidulafungin synthesis, this same reactivity is exploited to attach the side chain to the cyclic peptide nucleus. However, the presence of diethylamine represents a competitive pathway that diverts the intermediate away from the desired product. By synthesizing Impurity F independently, manufacturers gain a precise tool to quantify exactly how much of this diversion is occurring in their main process. This mechanistic clarity is essential for root cause analysis; if Impurity F levels spike in a production batch, engineers can immediately trace the issue back to the quality of the triethylamine base or the integrity of the activated ester intermediate, enabling rapid corrective actions.
How to Synthesize Anidulafungin Impurity Compound F Efficiently
The preparation of Anidulafungin Impurity F is designed for operational ease and reproducibility, making it accessible for both analytical laboratories and pilot-scale production facilities. The process begins by dissolving the intermediate SM-I in a selected organic solvent, with dimethyl sulfoxide and acetonitrile showing particularly favorable results in terms of solubility and reaction rate. Once the solution is prepared, diethylamine is added dropwise to manage the exotherm and ensure homogeneous mixing. The reaction mixture is then stirred at ambient temperatures, typically between 20°C and 30°C, for a duration of 3 to 5 hours, allowing sufficient time for the aminolysis to reach completion.
- Dissolve the anidulafungin intermediate compound SM-I in a suitable organic solvent such as dimethyl sulfoxide, acetonitrile, or acetone.
- Dropwise add diethylamine to the solution while maintaining the reaction temperature between 20°C and 30°C.
- Stir the mixture for 3 to 5 hours, then precipitate the product by adding water, followed by filtration and vacuum drying.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this patented synthesis method translates into tangible improvements in supply security and cost efficiency. The primary value proposition lies in the decoupling of reference standard production from the main API manufacturing schedule. Previously, the scarcity of Impurity F could bottleneck the entire quality control workflow, potentially delaying the release of valuable API batches. By establishing a dedicated, high-yield synthesis for this impurity, companies can build a strategic inventory of reference standards that is independent of API production volumes, thereby insulating the supply chain from fluctuations in main batch yields.
- Cost Reduction in Manufacturing: The new method drastically simplifies the production of the reference standard by eliminating the need for complex preparative HPLC purification. Traditional isolation methods often require large volumes of expensive solvents and significant column chromatography resources to separate trace impurities from the main product. In contrast, the patented route utilizes a precipitation workup where the addition of water causes the product to crash out of solution, allowing for simple filtration. This reduction in downstream processing steps significantly lowers the cost of goods sold for the reference standard, freeing up budget for other critical R&D activities while reducing the environmental footprint associated with solvent waste disposal.
- Enhanced Supply Chain Reliability: The raw materials required for this synthesis, specifically the SM-I intermediate and diethylamine, are commercially available and chemically stable. Unlike biological fermentation precursors which can suffer from batch-to-batch variability, these chemical inputs offer consistent quality and predictable lead times. This reliability ensures that the production of the impurity standard can be scaled up or down rapidly in response to demand without the long lead times associated with fermentative processes. Furthermore, the robustness of the reaction conditions means that the process can be easily transferred between different manufacturing sites or CDMO partners without extensive re-validation, providing flexibility in sourcing strategies.
- Scalability and Environmental Compliance: The process operates under mild conditions without the need for hazardous reagents or extreme temperatures, aligning well with modern green chemistry principles. The solvents used, such as acetonitrile and acetone, are widely recycled in pharmaceutical facilities, and the absence of heavy metal catalysts simplifies waste treatment protocols. From a scalability standpoint, the reaction kinetics are favorable for larger vessels, as the heat generation is manageable and the mixing requirements are standard. This ease of scale-up ensures that as regulatory requirements for impurity testing become more stringent globally, the supply of the necessary reference standards can grow in tandem without requiring massive capital investment in new specialized equipment.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Anidulafungin Impurity F. These insights are derived directly from the technical specifications and experimental data provided in patent CN112390723A, offering a clear understanding of how this technology integrates into existing pharmaceutical workflows.
Q: Why is Anidulafungin Impurity F critical for quality control?
A: Impurity F arises from the reaction of diethylamine contaminants in industrial triethylamine with the drug's side chain. Having a pure reference standard allows manufacturers to accurately quantify this specific impurity via HPLC, ensuring the final API meets strict regulatory safety limits.
Q: What is the advantage of the patented synthesis method over isolation?
A: Traditional isolation from reaction mixtures yields low quantities and poor purity. The patented method synthesizes Impurity F directly from SM-I and diethylamine, achieving yields over 95% and purity exceeding 98%, providing a reliable source for analytical standards.
Q: Is this synthesis scalable for commercial reference standard production?
A: Yes, the process uses common organic solvents like DMSO and acetonitrile and operates at mild temperatures (20-30°C). The simple workup involving water precipitation and filtration makes it highly amenable to kilogram-scale production without complex purification steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Anidulafungin Impurity F Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your final pharmaceutical product depends on the quality of every component in your supply chain, including the reference standards used for validation. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your demand for high-purity Anidulafungin Impurity F with consistency and speed. We adhere to stringent purity specifications and utilize rigorous QC labs to verify that every gram of material we supply meets the exacting standards required for regulatory submissions and routine quality control.
We invite you to collaborate with us to optimize your supply chain for echinocandin intermediates and impurities. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced synthesis capabilities can support your commitment to delivering safe and effective antifungal therapies to patients worldwide.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →