Advanced Synthesis of Azithromycin Impurity P: A Breakthrough in Pharmaceutical Reference Standards
Advanced Synthesis of Azithromycin Impurity P: A Breakthrough in Pharmaceutical Reference Standards
The pharmaceutical industry faces constant challenges in ensuring the safety and efficacy of antibiotic formulations, particularly regarding the identification and control of trace impurities. Patent CN109293722B, published in May 2020, addresses a critical gap in the quality control of Azithromycin, a widely used macrolide antibiotic. This patent details a novel preparation method for Azithromycin related substance, specifically identified as Impurity P, which was previously categorized as an unknown impurity in the European Pharmacopoeia. By successfully synthesizing and structurally characterizing this compound, the technology provides an essential reference standard that enables precise qualitative and quantitative analysis. This breakthrough not only fills a significant void in the Azithromycin impurity database but also significantly enhances medication safety by allowing manufacturers to strictly monitor and control this specific degradation product during the production process.
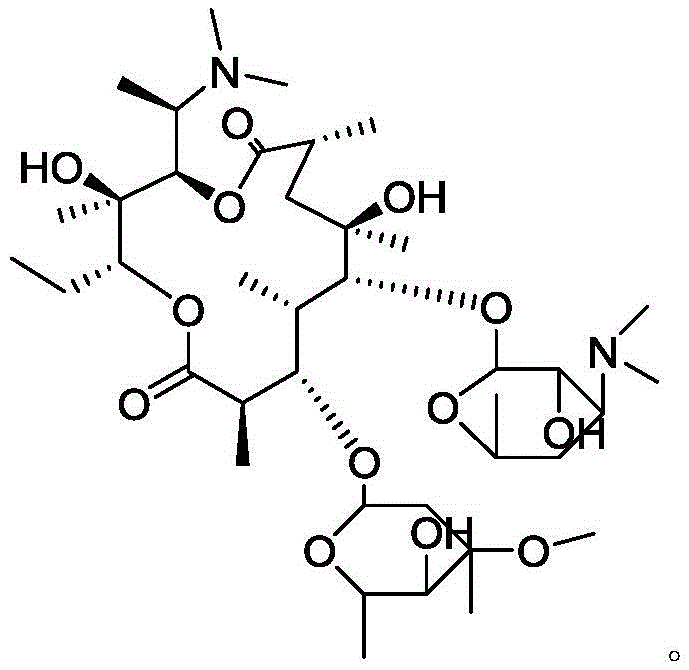
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the identification and acquisition of specific Azithromycin impurities have been fraught with difficulties, primarily because these substances often exist only in trace amounts within the final active pharmaceutical ingredient (API). Conventional approaches typically relied on the arduous task of isolating these impurities directly from large-scale fermentation broths or reaction mixtures where Azithromycin is produced. This isolation process is inherently inefficient, yielding minute quantities of the target compound that are often insufficient for comprehensive structural elucidation or for use as robust reference standards. Furthermore, without a pure sample of the impurity, analytical laboratories were forced to rely on relative retention times and estimated correction factors, which introduced uncertainty into the quality control process. The inability to definitively characterize Impurity P meant that its potential toxicity or impact on drug stability remained largely theoretical, posing a regulatory risk for manufacturers aiming to comply with stringent international pharmacopoeia standards such as the USP or EP.
The Novel Approach
The methodology described in patent CN109293722B represents a paradigm shift from passive isolation to active, targeted synthesis. Instead of scavenging for trace amounts of Impurity P, this approach utilizes erythromycin imino ether as a dedicated starting material to deliberately construct the impurity's molecular framework. The core innovation lies in the strategic manipulation of the hydrogenation step. By employing a specially pretreated platinum carbon catalyst, the process achieves a controlled reduction that halts at the desired intermediate stage rather than proceeding to the fully reduced Azithromycin. This is followed by a specific N-methylation reaction using formic acid and formaldehyde. This synthetic route allows for the generation of Impurity P in substantial quantities, with crude reaction contents reaching between 30% and 40%. This high initial concentration drastically simplifies the subsequent purification steps, making the production of high-purity reference standards commercially viable and technically reproducible on a scale that isolation methods could never achieve.
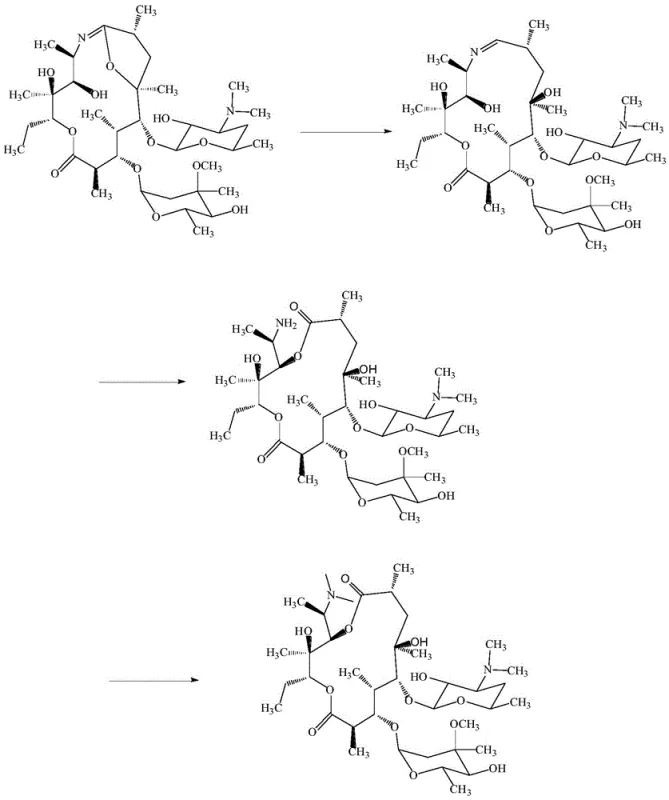
Mechanistic Insights into Modified Platinum Carbon Catalysis
The heart of this synthesis lies in the sophisticated modulation of the platinum carbon catalyst's activity. In standard hydrogenation processes, platinum carbon is highly active and would typically reduce the carbon-nitrogen double bond of the imino ether completely, leading directly to Azithromycin or other reduced byproducts. However, the patent reveals a clever mechanism wherein the catalyst is pretreated with specific metal ions, such as ferric ions (Fe3+), calcium ions (Ca2+), or copper ions (Cu2+). This pretreatment involves stirring the 5% platinum carbon in water with the metal salt for approximately thirty minutes before filtration and drying. The adsorption of these metal ions onto the catalyst surface appears to partially poison or deactivate the active sites. This controlled deactivation is crucial; it reduces the catalytic activity just enough to prevent the reduction of the C=N bond while still facilitating the necessary hydrogenation of other parts of the molecule or creating the specific acidic conditions required for imine hydrolysis. Consequently, the C=N bond undergoes hydrolysis under acidic conditions to form an amino group, which is then available for the subsequent methylation.
Following the hydrogenation, the resulting intermediate undergoes an Eschweiler-Clarke type N-methylation reaction. The process involves dissolving the dried hydrogenation product in acetone and reacting it with formic acid and formaldehyde at a controlled pH of 5-6 and a temperature of 40-50°C. This step introduces two methyl groups to the nitrogen atom, finalizing the structure of Impurity P. The precision of this mechanism ensures that the stereochemistry of the macrocyclic ring is preserved while specifically modifying the amine functionality. The result is a molecule that closely mimics the degradation product found in aged Azithromycin samples but is produced through a deterministic chemical pathway. This mechanistic understanding allows for fine-tuning of reaction parameters, such as maintaining hydrogen pressure at 1.0-1.2MPa and temperatures at 55-60°C, to maximize the yield of the target impurity while minimizing the formation of unrelated side products.
How to Synthesize Azithromycin Impurity P Efficiently
The synthesis of Azithromycin Impurity P requires precise adherence to the patented protocol to ensure the correct structural formation and high purity. The process begins with the critical pretreatment of the catalyst, followed by a controlled hydrogenation and a subsequent methylation step. The reaction conditions, including pH, pressure, and temperature, must be strictly monitored to favor the formation of the impurity over the parent drug. Once the crude product is obtained, advanced chromatographic techniques are employed to isolate the target compound from the reaction matrix. For a detailed breakdown of the standardized operating procedures and specific reagent grades required for this synthesis, please refer to the technical guide below.
- Pretreat 5% platinum carbon catalyst by stirring in water with metal ions (e.g., Fe3+) for 30 minutes to modulate catalytic activity.
- Perform hydrogenation of erythromycin imino ether at 1.0-1.2MPa and 55-60°C using the treated catalyst, followed by N-methylation with formic acid and formaldehyde.
- Purify the crude product via liquid preparative chromatography using a C18 column to achieve purity levels exceeding 95%.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors in the pharmaceutical sector, the ability to source high-purity reference standards is not merely a technical requirement but a strategic necessity for regulatory compliance. The synthesis method outlined in this patent offers significant commercial advantages by transforming the production of Azithromycin Impurity P from a scarce, isolation-dependent resource into a reliably manufacturable commodity. By shifting to a synthetic route, suppliers can guarantee consistent batch-to-batch quality and availability, eliminating the volatility associated with extracting trace impurities from variable biological sources. This stability in supply is crucial for pharmaceutical companies that must validate their analytical methods and maintain continuous quality control operations without interruption. Furthermore, the ability to produce this impurity in larger quantities supports the growing global demand for generic Azithromycin, where rigorous impurity profiling is mandatory for market approval.
- Cost Reduction in Manufacturing: The transition from isolation to synthesis fundamentally alters the cost structure of producing reference standards. Isolation methods are labor-intensive and suffer from extremely low yields, driving up the cost per gram exponentially. In contrast, this synthetic route utilizes readily available starting materials like erythromycin imino ether and common reagents such as formic acid and formaldehyde. The elimination of complex extraction processes and the ability to generate the crude impurity at concentrations of 30-40% significantly reduces the volume of solvents and consumables required for purification. Additionally, the use of recoverable catalysts and standard reactor equipment avoids the need for specialized, expensive processing infrastructure, leading to substantial overall cost savings in the manufacturing of these critical quality control materials.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the deterministic nature of this chemical synthesis. Unlike biological isolation, which is subject to the fluctuations of fermentation yields and the complexity of downstream processing, this chemical route can be scaled predictably. The raw materials, including erythromycin derivatives and metal salts for catalyst treatment, are commercially available from multiple global suppliers, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions, which tolerate standard industrial parameters for pressure and temperature, ensures that production can be maintained consistently even during periods of high demand. This reliability allows pharmaceutical manufacturers to secure long-term contracts for reference standards, ensuring that their quality control laboratories never face shortages that could delay product release.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing unit operations such as autoclave hydrogenation and crystallization that are easily transferred from laboratory to pilot and commercial scales. The purification step employs preparative liquid chromatography, a technique that can be scaled up using larger columns and automated systems to handle increased throughput. From an environmental perspective, the synthesis avoids the use of hazardous heavy metal catalysts that require complex removal steps; instead, it uses platinum carbon which can be filtered and potentially regenerated. The solvent systems involved, primarily methanol, acetone, and water, are well-understood and can be efficiently recovered and recycled in a modern facility. This alignment with green chemistry principles simplifies waste management and ensures compliance with increasingly stringent environmental regulations governing pharmaceutical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Azithromycin Impurity P. These insights are derived directly from the experimental data and structural analysis provided in the patent documentation. Understanding these details is vital for R&D teams integrating this reference standard into their analytical workflows and for procurement specialists evaluating supplier capabilities.
Q: Why is the pretreatment of platinum carbon critical in this synthesis?
A: The pretreatment with metal ions like Fe3+ partially deactivates the platinum carbon catalyst. This controlled reduction in activity prevents the complete hydrogenation of the carbon-nitrogen double bond, allowing for the selective formation of the specific imine hydrolysis product required for Impurity P.
Q: What is the primary application of the synthesized Azithromycin Impurity P?
A: The synthesized substance serves as a critical reference standard for qualitative and quantitative analysis. It allows pharmaceutical manufacturers to accurately identify and quantify this specific unknown impurity (relative retention time 0.92) in final Azithromycin products to meet pharmacopoeia standards.
Q: How does this synthetic route improve upon traditional isolation methods?
A: Traditional methods often struggle to isolate trace impurities from complex fermentation broths in sufficient quantities. This patented synthetic route starts from erythromycin imino ether, enabling the deliberate production of the impurity in significant yields (30-40% crude content), which simplifies downstream purification and ensures a stable supply.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Azithromycin Impurity P Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality reference standards play in the global pharmaceutical supply chain. Our team of expert chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of international regulatory bodies. We are committed to delivering Azithromycin Impurity P with stringent purity specifications, utilizing our state-of-the-art rigorous QC labs to verify every batch against the structural data confirmed in patent CN109293722B. Our capability to reproduce the specific catalytic hydrogenation and methylation steps ensures that the impurity profile of our reference standards matches the authentic substance found in pharmaceutical products, providing you with the confidence needed for accurate method validation.
We invite you to collaborate with us to optimize your quality control processes and ensure the safety of your antibiotic formulations. Our technical sales team is ready to provide a Customized Cost-Saving Analysis tailored to your specific consumption volumes and purity requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. By partnering with us, you gain access to a reliable supply of critical impurities that supports your commitment to excellence in pharmaceutical manufacturing and patient safety.