Advanced Chiral Tetrahydrofuran Synthesis: Scalable Routes for Complex API Intermediates
Introduction to High-Selectivity Tetrahydrofuran Construction
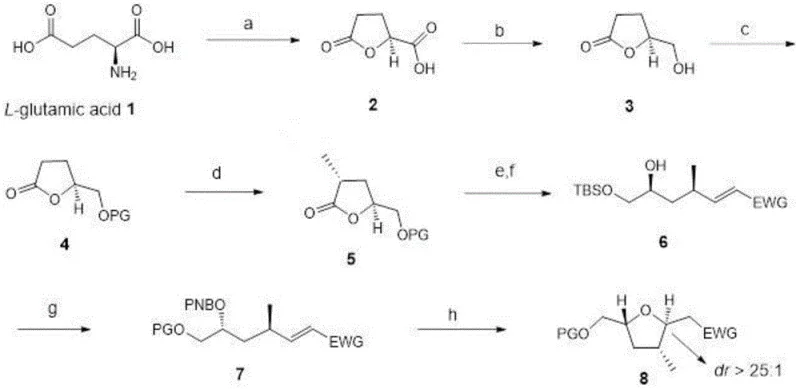
The construction of multi-chiral center tetrahydrofuran rings represents a formidable challenge in modern organic synthesis, particularly given their prevalence as core skeletons in bioactive natural products such as Chagosensine and Amphinolides. Patent CN110305159B discloses a groundbreaking methodology that addresses the longstanding difficulties in achieving high regioselectivity and stereoselectivity for these complex architectures. By leveraging a chiral pool strategy starting from the abundant and inexpensive L-glutamic acid, this invention offers a robust pathway that circumvents the pitfalls of earlier transition-metal catalyzed or base-promoted approaches. The significance of this technology lies not only in its academic elegance but in its practical applicability for the commercial scale-up of complex pharmaceutical intermediates. For R&D directors and process chemists, the ability to access these scaffolds with predictable stereochemistry is paramount for accelerating drug discovery pipelines.
This novel synthetic route effectively integrates eight distinct transformation steps, ranging from diazotization to Mitsunobu inversion, into a cohesive and efficient sequence. The method ensures that the critical 2,5-trans-tetrahydrofuran substitution pattern is established with exceptional fidelity, overcoming the diastereomeric mixture issues that have plagued previous attempts. As a reliable pharmaceutical intermediate supplier, understanding such proprietary routes allows us to offer clients superior supply chain security for high-value chiral building blocks. The following analysis details the mechanistic nuances and commercial implications of this patented technology.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies for constructing 3-substituted 2,5-trans-tetrahydrofurans have historically been fraught with significant technical hurdles that impede efficient manufacturing. For instance, the Michael addition reactions promoted by TBAF, as reported by the William R. Roush team, often result in a diastereomeric ratio (dr) of merely 10:1. This lack of stereocontrol necessitates cumbersome purification processes to isolate the desired isomer, drastically reducing the overall throughput and increasing the cost of goods sold. Furthermore, alternative approaches utilizing DBU promotion, such as those by the Christopher D. Spilling group, suffer from competing side reactions where the hydroxyl protecting groups undergo undesirable migration. Specifically, the transfer of TBS groups under basic conditions leads to yields as low as 32% for the target configuration. These inefficiencies create substantial bottlenecks in cost reduction in API manufacturing, as low yields and difficult separations translate directly into higher raw material consumption and extended production timelines.
The Novel Approach
In stark contrast, the method disclosed in CN110305159B introduces a streamlined sequence that capitalizes on the intrinsic chirality of L-glutamic acid to dictate stereochemical outcomes. By initiating the synthesis with a diazotization and ring-closing event, the process locks in the initial stereocenters with high fidelity before proceeding through a series of functional group manipulations. The use of a Domino strategy and one-pot methods for hydrolysis and intramolecular asymmetric Michael addition ring closure significantly simplifies the operational complexity. This approach effectively eliminates the TBS transfer issues seen in prior art, ensuring that the protecting groups remain intact until the intended deprotection step. The result is a synthesis characterized by mild conditions and high stereoselectivity, providing a reliable source of high-purity pharmaceutical intermediates that meet the stringent quality standards required for clinical applications.
Mechanistic Insights into Chiral Pool Strategy and Mitsunobu Inversion
The core strength of this synthesis lies in its meticulous orchestration of stereochemical events, beginning with the conversion of L-glutamic acid into a lactone intermediate. The initial diazotization step serves a dual purpose: it removes the amino group while simultaneously triggering an intramolecular cyclization to form the five-membered lactone ring. This step is crucial as it preserves the chirality at the C4 position (relative to the glutamic acid backbone), which eventually becomes a key stereocenter in the final tetrahydrofuran product. Subsequent reduction of the carboxyl group using borane-dimethyl sulfide complex proceeds with high chemoselectivity, leaving the lactone carbonyl untouched. The protection of the resulting primary alcohol with a tert-butyldimethylsilyl (TBS) group provides the necessary stability for the subsequent alpha-alkylation. This alkylation step, mediated by LiHMDS at cryogenic temperatures (-78°C), introduces the methyl group at the alpha-position with excellent diastereocontrol, driven by the existing chiral environment of the lactone ring.
The latter stages of the synthesis employ a sophisticated sequence of reduction, olefination, and inversion to finalize the ring architecture. The reduction of the lactone to a lactol followed by a Wittig reaction installs the requisite carbon chain with an electron-withdrawing group, setting the stage for ring closure. A pivotal moment in the mechanism is the Mitsunobu reaction, which utilizes triphenylphosphine and DIAD in the presence of p-nitrobenzoic acid. This reaction facilitates an SN2-type inversion of configuration at the secondary alcohol center, ensuring the correct relative stereochemistry between the substituents. Finally, the removal of the protecting groups under basic conditions triggers a spontaneous intramolecular cyclization. This cascade effectively constructs the target 2,5-trans-tetrahydrofuran ring with a dr value exceeding 25:1, demonstrating the power of combining chiral pool starting materials with classic name reactions to solve complex stereochemical problems.
How to Synthesize Multi-Chiral-Center Tetrahydrofuran Efficiently
The execution of this synthesis requires careful attention to reaction conditions, particularly temperature control during the alkylation and reduction steps to maintain stereochemical integrity. The process begins with the preparation of the lactone scaffold from L-glutamic acid, followed by sequential functionalization to build the carbon framework. Each step has been optimized to maximize yield and minimize byproduct formation, ensuring a smooth progression towards the final target. For process chemists looking to implement this route, the availability of detailed experimental procedures is essential for successful technology transfer and scale-up. The following guide outlines the standardized operational protocol derived from the patent examples.
- Perform diazotization and ring closure on L-glutamic acid using NaNO2 and HCl to form lactone intermediate 2.
- Reduce the carboxyl group of intermediate 2 with BH3·SMe2, protect the resulting alcohol with TBSCl to obtain intermediate 4.
- Execute alpha-alkylation with MeI/LiHMDS, followed by DIBAL reduction and Wittig reaction to install the side chain.
- Complete the synthesis via Mitsunobu reaction with p-nitrobenzoic acid followed by base-mediated deprotection and cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this synthetic route offers transformative benefits regarding cost efficiency and supply reliability. Traditional methods for generating multi-chiral tetrahydrofurans often rely on expensive chiral catalysts or resolution techniques that inherently waste 50% of the material. By utilizing L-glutamic acid, a commodity chemical available in massive quantities from fermentation processes, this method anchors the supply chain in a stable and cost-effective raw material base. The elimination of transition metal catalysts further reduces the burden on downstream processing, as there is no need for costly heavy metal scavenging steps to meet regulatory limits for residual metals in pharmaceutical ingredients. This simplification of the purification train translates directly into significant operational expenditure savings.
- Cost Reduction in Manufacturing: The high yields observed in key steps, such as the 92% yield in the initial lactone formation and 89% in the final cyclization, contribute to a highly atom-economical process. Unlike prior art methods that suffered from yields as low as 32% due to side reactions, this robust pathway minimizes raw material waste. The avoidance of specialized chiral reagents in favor of a chiral pool starting material drastically lowers the input cost per kilogram of the final intermediate. Furthermore, the use of standard reagents like TBSCl, LiHMDS, and common oxidants/reductants ensures that the bill of materials remains competitive, facilitating substantial cost savings in the overall manufacturing budget without compromising on quality.
- Enhanced Supply Chain Reliability: The reliance on L-glutamic acid as the starting material mitigates supply risk, as this amino acid is produced globally at industrial scales for the food and feed industries. This abundance ensures that fluctuations in the availability of exotic chiral precursors do not impact production schedules. Additionally, the operational simplicity of the reaction steps—many of which can be performed at ambient or easily achievable cryogenic temperatures—reduces the dependency on specialized reactor configurations. This flexibility allows for manufacturing across a wider range of facilities, thereby diversifying the supply base and reducing lead time for high-purity pharmaceutical intermediates in the event of regional disruptions.
- Scalability and Environmental Compliance: The synthetic route is designed with scalability in mind, utilizing solvents and reagents that are manageable on a multi-ton scale. The high stereoselectivity (dr > 25:1) means that less solvent and energy are consumed in chromatographic separations compared to low-selectivity routes. From an environmental standpoint, the absence of toxic heavy metals and the use of relatively benign byproducts align with green chemistry principles. This compliance simplifies the waste treatment process and reduces the environmental footprint of the manufacturing site, making it easier to obtain the necessary regulatory approvals for commercial production in strict jurisdictions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this high-selectivity synthesis method. These insights are derived directly from the comparative data and experimental results presented in the patent documentation, providing clarity on the method's superiority over existing technologies. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this intermediate into their drug development programs.
Q: What are the advantages of this tetrahydrofuran synthesis method over prior art?
A: Unlike previous methods suffering from low diastereoselectivity (dr 10:1) or protecting group migration issues, this route utilizes a chiral pool strategy from L-glutamic acid to achieve high stereoselectivity (dr > 25:1) and avoids TBS transfer side reactions.
Q: Is this process suitable for large-scale manufacturing of API intermediates?
A: Yes, the process features mild reaction conditions, simple operational steps, and high yields (e.g., 92% in the initial cyclization), making it highly amenable to commercial scale-up for complex pharmaceutical intermediates.
Q: What is the key stereochemical control strategy employed?
A: The synthesis leverages the inherent chirality of L-glutamic acid and employs a strategic Mitsunobu inversion to establish the critical 2,5-trans configuration with a 3-position quaternary carbon center.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tetrahydrofuran Derivative Supplier
The technological advancements described in CN110305159B highlight the immense potential of chiral pool synthesis in delivering complex scaffolds for next-generation therapeutics. At NINGBO INNO PHARMCHEM, we possess the technical expertise to adapt and optimize such sophisticated routes for industrial application. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to pilot plant is seamless. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every batch of tetrahydrofuran derivative meets the exacting standards required by global regulatory bodies.
We invite pharmaceutical partners to collaborate with us to leverage this efficient synthesis for their specific API needs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your project volume. We encourage you to reach out for specific COA data and route feasibility assessments to determine how this high-selectivity method can enhance your supply chain resilience and accelerate your time to market.