Optimizing Lenvatinib Mesylate Production: A Technical Breakthrough in High-Purity Crystallization
The pharmaceutical industry continuously demands more efficient and robust purification strategies for complex oncology agents, and patent CN110818634B represents a significant leap forward in the manufacturing of Lenvatinib Mesylate. This intellectual property discloses a novel refining method that addresses the critical bottlenecks of yield loss and impurity retention found in legacy processes. By shifting away from traditional单一 solvent crystallization towards a sophisticated mixed-solvent system involving ethylene glycol ethers, the technology achieves an HPLC purity exceeding 99.80% while maintaining single impurity levels below 0.10%. For R&D directors and process chemists, this breakthrough offers a viable pathway to overcome the thermodynamic limitations of previous methods, ensuring that the final Active Pharmaceutical Ingredient (API) meets the rigorous safety profiles required for tyrosine kinase inhibitors. The strategic implementation of this refining protocol not only enhances product quality but also aligns with the broader industry goals of green chemistry and waste reduction.
As a reliable API intermediate supplier, understanding the structural nuances of Lenvatinib Mesylate is paramount. The molecule, characterized by its quinoline core and urea linkage, presents specific challenges during isolation due to the presence of closely related synthetic byproducts. The patent highlights that conventional purification techniques often fail to adequately separate these structural analogs, leading to batches that require multiple recrystallizations or extensive chromatographic polishing. This new approach leverages specific solvation interactions to selectively precipitate the target mesylate salt while keeping detrimental impurities in the mother liquor. Such precision is essential for maintaining the therapeutic index of the drug and minimizing the risk of adverse effects in patients undergoing treatment for differentiated thyroid cancer.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, as detailed in documents like CN200480036184.1 and various journal publications, typically rely on crystallizing crude Lenvatinib Mesylate from单一 solvents such as methanol or ethanol, or binary mixtures involving dichloromethane, N,N-dimethylformamide (DMF), and acetonitrile. These traditional systems suffer from inherent thermodynamic deficiencies where the solubility gap between the desired product and key impurities is insufficiently wide. Consequently, the refining yield in these legacy processes is notoriously low, often ranging between 50% and 70%, which translates to substantial material loss and increased cost of goods sold (COGS). Furthermore, the residual impurity content, particularly Impurity II and Impurity III, frequently remains above acceptable thresholds, necessitating repeated processing cycles that degrade overall throughput and extend production lead times significantly.
The Novel Approach
In stark contrast, the novel approach defined in CN110818634B introduces a tailored solvent engineering strategy that fundamentally alters the crystallization landscape. By employing a mixed solvent system comprising ethylene glycol dimethyl carbonate (or mono-ethers) combined with C1-C4 alkyl alcohols or alkyl esters, the process creates an optimal environment for selective nucleation. This specific chemical environment allows for the dissolution of the crude material at elevated temperatures (45°C to 80°C) followed by controlled cooling to induce high-purity crystal growth. The result is a dramatic improvement in refining yield, consistently achieving between 81% and 87%, alongside an HPLC purity greater than 99.80%. This shift represents a paradigm change in cost reduction in pharmaceutical manufacturing, as it maximizes the output from every kilogram of crude input while minimizing solvent consumption and waste generation.
Mechanistic Insights into Solvent-Mediated Crystallization and Impurity Rejection
The efficacy of this refining method lies in the precise modulation of solvent polarity and hydrogen bonding capabilities. Ethylene glycol ethers possess unique solvation properties that interact differently with the polar urea and quinoline functionalities of Lenvatinib compared to the non-polar or less polar impurities. When mixed with alcohols like methanol or ethanol, or esters like ethyl acetate, the solvent system fine-tunes the saturation point of the API. During the cooling phase, typically from roughly 55°C down to 5°C to 15°C, the target molecule reaches supersaturation and precipitates out in a highly ordered crystalline lattice. Meanwhile, structurally similar impurities, which might co-crystallize in simpler solvent systems, remain solvated in the mother liquor due to the specific steric and electronic mismatches introduced by the glycol ether component. This mechanism ensures that the crystal lattice is energetically unfavorable for the inclusion of foreign molecules.
Furthermore, the control of impurity profiles is critical for regulatory compliance and patient safety. The patent specifically identifies Impurity II and Impurity III as major concerns in the crude product, often arising from incomplete reactions or side reactions during the synthesis of the quinoline-urea scaffold. 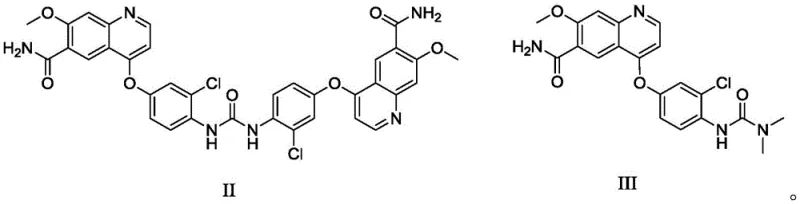 The new crystallization protocol demonstrates a remarkable capacity to reject these specific contaminants, reducing their levels to below 0.10% individually. This level of control is achieved not just by the solvent choice but also by optimizing the cooling rate (1°C/hr to 50°C/hr) and crystallization time (1 to 5 hours). Such parameters prevent the rapid, disordered precipitation that often traps impurities within the crystal matrix, thereby ensuring a high-purity API intermediate that requires minimal downstream processing before formulation.
The new crystallization protocol demonstrates a remarkable capacity to reject these specific contaminants, reducing their levels to below 0.10% individually. This level of control is achieved not just by the solvent choice but also by optimizing the cooling rate (1°C/hr to 50°C/hr) and crystallization time (1 to 5 hours). Such parameters prevent the rapid, disordered precipitation that often traps impurities within the crystal matrix, thereby ensuring a high-purity API intermediate that requires minimal downstream processing before formulation.
How to Synthesize Lenvatinib Mesylate Efficiently
Implementing this refined purification protocol requires careful attention to solvent ratios and thermal profiles to replicate the high yields and purity reported in the patent data. The process begins with the selection of high-quality crude Lenvatinib Mesylate, typically possessing an initial purity of 95.0% to 99.0%, which is then subjected to the specialized solvent treatment. The detailed standardized synthesis steps involve dissolving the crude material in the specific glycol ether/alcohol mixture at controlled temperatures, followed by a gradual cooling ramp to facilitate the growth of large, pure crystals. For process engineers and plant managers, adhering to these specific parameters is crucial for scaling the technology from laboratory benchtop to commercial production vessels without losing the selectivity benefits observed in the initial trials.
- Dissolve crude Lenvatinib Mesylate in a heated mixture of ethylene glycol dimethyl ether and an alkyl alcohol solvent.
- Cool the solution gradually to induce controlled crystallization, separating the pure product from impurities.
- Filter, wash with cold solvent, and vacuum dry to obtain Lenvatinib Mesylate with >99.8% HPLC purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this advanced refining technology offers tangible strategic benefits that extend beyond mere technical specifications. The primary advantage lies in the substantial increase in process yield, which directly correlates to a reduction in raw material consumption per unit of finished API. By boosting recovery rates from the historical average of roughly 60% to over 85%, manufacturers can significantly lower the effective cost of production without compromising on quality standards. This efficiency gain is particularly vital in the context of oncology drugs, where starting materials can be expensive and supply chains are often fragile. Additionally, the simplification of the purification workflow eliminates the need for multiple recrystallization steps or complex chromatographic separations, thereby reducing solvent usage, energy consumption, and overall processing time.
- Cost Reduction in Manufacturing: The elimination of inefficient purification cycles leads to direct savings in operational expenditures. By achieving high purity in a single crystallization step, the process reduces the burden on utility systems and labor, allowing for a leaner manufacturing footprint. The use of commercially available and relatively inexpensive solvents like ethylene glycol ethers and ethanol further contributes to a favorable cost structure, making the final API more competitive in the global marketplace.
- Enhanced Supply Chain Reliability: Higher yields and robust process parameters translate to more predictable production schedules and shorter lead times. The ability to consistently produce material that meets strict purity specifications (>99.8%) reduces the risk of batch failures and rejections, ensuring a steady flow of product to downstream formulation partners. This reliability is essential for maintaining continuous supply to patients and avoiding costly disruptions in the pharmaceutical distribution network.
- Scalability and Environmental Compliance: The method is explicitly designed for industrial production, utilizing standard unit operations such as heating, cooling, filtration, and vacuum drying. This compatibility with existing infrastructure facilitates rapid scale-up from pilot plants to multi-ton commercial facilities. Moreover, the reduced solvent volume and improved material efficiency align with modern environmental, social, and governance (ESG) goals, minimizing the ecological impact of API manufacturing and simplifying waste management protocols.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this refining technology. They are derived from the specific experimental data and comparative examples provided in the patent documentation, offering clarity on how this method outperforms traditional approaches in real-world scenarios. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this process into their existing manufacturing portfolios.
Q: What is the primary advantage of the new refining method over conventional methanol crystallization?
A: The new method utilizes a specific mixed solvent system (glycol ethers and alcohols/esters) that significantly improves the solubility differential between the API and key impurities (II and III), resulting in higher yields (81-87% vs 50-70%) and superior purity (>99.8%).
Q: Which specific impurities are effectively removed by this process?
A: The process is specifically optimized to reduce Impurity II and Impurity III to levels below 0.10% each, meeting stringent pharmacopoeial standards for oncology drugs.
Q: Is this purification method suitable for industrial scale-up?
A: Yes, the patent explicitly states the method is suitable for industrial production due to its operational simplicity, high recovery rates, and the use of commercially available, scalable solvent systems.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Lenvatinib Mesylate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity intermediates in the development of life-saving oncology therapies. Our team of expert chemists has extensively analyzed the technological advancements presented in patent CN110818634B and possesses the capability to implement these sophisticated purification strategies at scale. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive material that not only meets but exceeds stringent purity specifications. Our state-of-the-art rigorous QC labs are equipped to verify the absence of critical impurities like Impurity II and III, guaranteeing the safety and efficacy of the supply we provide to our global partners.
We invite pharmaceutical companies and contract research organizations to collaborate with us to leverage this cutting-edge refining technology for their Lenvatinib projects. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis that quantifies the potential economic benefits of switching to this high-yield process. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our commitment to innovation can drive value and efficiency in your supply chain.