Scalable Synthesis of Terbutaline Sulfate Impurity B for Global Pharmaceutical Quality Control
The pharmaceutical industry relies heavily on precise impurity profiling to ensure the safety and efficacy of active pharmaceutical ingredients (APIs). A critical challenge in this domain is the availability of high-purity reference standards for specific degradation products or process-related impurities. Patent CN115010665A addresses this gap by introducing a novel, efficient method for synthesizing Terbutaline Sulfate Impurity B. Terbutaline sulfate is a widely used selective beta-2 adrenergic receptor agonist for treating asthma and other respiratory conditions, and strict regulatory bodies require rigorous control over its impurity profile. Historically, obtaining sufficient quantities of Impurity B for analytical validation has been problematic because it forms only in trace amounts during standard API manufacturing. This new technical breakthrough offers a dedicated, five-step synthetic pathway that transforms readily available starting materials into the target impurity with exceptional efficiency. By establishing a reliable supply of this critical reference standard, manufacturers can significantly enhance their quality control protocols, ensuring that every batch of Terbutaline Sulfate meets the stringent purity specifications demanded by global pharmacopoeias.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the acquisition of specific impurity standards like Terbutaline Sulfate Impurity B has been fraught with logistical and technical difficulties. In conventional API manufacturing processes, Impurity B is an incidental byproduct generated in minute quantities, often buried within a complex matrix of the main product and other side reactions. Attempting to isolate this impurity from the crude reaction mixture requires extensive and costly purification techniques, such as preparative HPLC or repeated recrystallization, which often result in negligible recovery rates. Furthermore, the structural similarity between the impurity and the parent drug makes separation incredibly challenging, leading to reference standards that may lack the necessary purity for accurate quantitative analysis. This scarcity forces quality control laboratories to rely on external suppliers with long lead times or to accept lower-quality standards that compromise the accuracy of their stability studies. The inability to consistently source high-purity Impurity B creates a bottleneck in the regulatory approval process for generic manufacturers and complicates the ongoing stability monitoring of the drug product throughout its shelf life.
The Novel Approach
The methodology disclosed in patent CN115010665A represents a paradigm shift by treating the impurity as a primary target rather than a waste product. Instead of relying on isolation, this approach constructs the molecule de novo through a logical sequence of esterification, substitution, cyclization, hydrolysis, and reduction. This dedicated synthesis allows for the optimization of each step specifically for the formation of the impurity structure, bypassing the thermodynamic constraints that limit its formation in the main API process. The route utilizes cost-effective starting materials like 2-bromo-3',5'-dihydroxyacetophenone and employs standard organic transformations that are well-understood and easily controlled. By decoupling the production of the impurity standard from the API manufacturing line, this method ensures a consistent, independent supply chain. The result is a robust process capable of delivering the target compound with a total yield exceeding 50%, a figure that is remarkably high for a multi-step synthesis of a complex heterocyclic system, thereby solving the supply scarcity issue definitively.
Mechanistic Insights into the Five-Step Synthetic Route
The chemical elegance of this synthesis lies in its strategic use of protecting groups and sequential functionalization to build the tetrahydroisoquinoline core characteristic of Terbutaline derivatives. The process initiates with the protection of the phenolic hydroxyl groups on the starting acetophenone via esterification with acetic anhydride. This step is crucial as it prevents unwanted side reactions at the phenolic positions during the subsequent nucleophilic substitution with tert-butylamine. Once the amine is installed, the molecule undergoes a cyclization reaction, likely a Mannich-type condensation using formaldehyde or paraformaldehyde, which closes the ring to form the nitrogen-containing heterocycle. This cyclization step is the key structural determinant that differentiates the linear precursor from the final bicyclic impurity. Following ring closure, the acetate protecting groups are removed through alkaline hydrolysis, regenerating the free phenolic hydroxyls essential for the biological activity and structural identity of the molecule. The final step involves the reduction of the ketone functionality to a secondary alcohol, completing the transformation into Terbutaline Sulfate Impurity B.
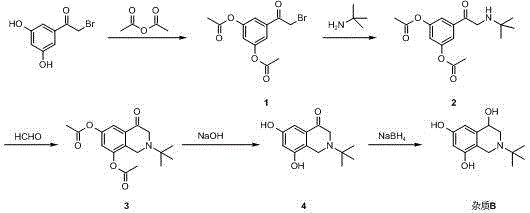
Controlling the stereochemistry and regioselectivity throughout this pathway is vital for ensuring the final product matches the authentic impurity found in the API. The patent details specific conditions, such as the use of sodium borohydride or lithium aluminum hydride for the final reduction, which allows for precise control over the reaction kinetics. The choice of solvents, ranging from dichloromethane for esterification to methanol or ethanol for substitution and hydrolysis, is optimized to maximize solubility and reaction rates while minimizing byproduct formation. For instance, the cyclization step is performed under mild conditions, often at room temperature or with slight heating, which prevents the degradation of the sensitive intermediate structures. The hydrolysis step uses aqueous alkali solutions to efficiently cleave the ester bonds without affecting the newly formed amine or alcohol functionalities. This careful orchestration of reaction conditions ensures that the final product is not only structurally correct but also possesses the high purity required for use as an analytical reference standard, effectively eliminating the need for complex downstream purification.
How to Synthesize Terbutaline Sulfate Impurity B Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires strict adherence to the optimized parameters outlined in the patent to achieve the reported yields and purity levels. The process is designed to be operationally simple, avoiding the need for exotic catalysts or extreme pressure conditions, which facilitates easy technology transfer. Operators should focus on maintaining precise stoichiometric ratios, particularly during the substitution and cyclization steps, to prevent the formation of oligomeric byproducts. The workup procedures, involving standard extraction and crystallization techniques, are straightforward but critical for removing inorganic salts and residual solvents. For detailed operational parameters, including specific temperatures, reaction times, and molar ratios for each of the five steps, please refer to the standardized guide below.
- Esterification of 2-bromo-3',5'-dihydroxyacetophenone with acetic anhydride to protect phenolic hydroxyl groups.
- Nucleophilic substitution with tert-butylamine followed by cyclization with formaldehyde to form the tetrahydroisoquinoline core.
- Hydrolysis of acetate protecting groups and final reduction of the ketone to yield the target impurity standard.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers significant strategic advantages beyond mere technical feasibility. The primary benefit is the drastic simplification of the sourcing strategy for critical quality control materials. By moving from an isolation-dependent model to a dedicated synthesis model, companies can secure a stable, long-term supply of Impurity B that is immune to fluctuations in API production volumes. This independence reduces the risk of supply chain disruptions that could halt quality testing and delay product release. Furthermore, the use of commodity chemicals as starting materials means that the cost of goods sold for this impurity standard is significantly lower compared to custom synthesis services that charge premiums for difficult isolations. The robustness of the five-step process also implies a lower rate of batch failures, ensuring consistent availability and reducing the administrative burden of managing multiple suppliers for niche reference standards.
- Cost Reduction in Manufacturing: The economic viability of this method stems from the high overall yield and the use of inexpensive, commercially available reagents. Unlike traditional methods that might require tons of API crude to isolate grams of impurity, this route builds the molecule efficiently from cheap precursors. The elimination of expensive preparative chromatography steps, which are typically required for isolation, further drives down processing costs. Additionally, the mild reaction conditions reduce energy consumption and equipment wear, contributing to a leaner manufacturing cost structure. These savings can be passed down to the end-user, making high-quality reference standards more accessible for routine quality control testing without compromising budget allocations.
- Enhanced Supply Chain Reliability: Supply chain resilience is greatly improved because the raw materials for this synthesis are not specialized intermediates but rather bulk chemicals with established global supply networks. This reduces the lead time associated with procuring starting materials and minimizes the risk of single-source dependency. The scalability of the process means that production can be ramped up quickly to meet sudden spikes in demand, such as during regulatory audits or new product filings. By internalizing or partnering with a supplier who utilizes this specific patent-protected route, organizations can guarantee the continuity of their quality control operations, ensuring that no batch of Terbutaline Sulfate goes untested due to a lack of reference standards.
- Scalability and Environmental Compliance: From an environmental and safety perspective, this route is superior to many alternative synthetic pathways. The reactions proceed under mild conditions, avoiding the use of highly hazardous reagents or extreme temperatures that pose safety risks at scale. The solvents used, such as ethanol and methanol, are relatively benign and easily recoverable, facilitating effective waste management and recycling programs. The high atom economy of the cyclization and reduction steps minimizes waste generation, aligning with green chemistry principles. This environmental compatibility simplifies the permitting process for manufacturing facilities and reduces the costs associated with hazardous waste disposal, making the entire operation more sustainable and compliant with increasingly strict environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Terbutaline Sulfate Impurity B. These answers are derived directly from the technical specifications and experimental data provided in the patent literature, ensuring accuracy and relevance for industry professionals. Understanding these details is crucial for R&D teams planning to implement this standard in their analytical workflows and for procurement teams evaluating supplier capabilities. The clarity provided here aims to remove ambiguity regarding the synthesis quality and its suitability for regulatory purposes.
Q: Why is synthesizing Terbutaline Sulfate Impurity B challenging?
A: Impurity B is typically generated only in trace amounts during the main API synthesis, making isolation and purification for reference standards extremely difficult and inefficient without a dedicated synthetic route.
Q: What is the overall yield of the patented synthesis method?
A: The patented method described in CN115010665A achieves a total yield of greater than 50% over five steps, which is significantly higher than isolation methods from crude API mixtures.
Q: Is this synthesis route suitable for large-scale production?
A: Yes, the process utilizes readily available raw materials and mild reaction conditions (e.g., room temperature esterification, moderate heating for substitution), making it highly amenable to commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Terbutaline Sulfate Impurity B Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your pharmaceutical products depends on the quality of your reference standards. As a leading CDMO and fine chemical manufacturer, we have successfully integrated advanced synthetic methodologies, such as the one described in patent CN115010665A, into our production capabilities. Our facility is equipped to handle the commercial scale-up of complex pharmaceutical intermediates, with extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We maintain stringent purity specifications and operate rigorous QC labs to ensure that every gram of Terbutaline Sulfate Impurity B we supply meets the highest international standards. Our commitment to technical excellence ensures that you receive a product that is not only chemically pure but also fully characterized and ready for immediate use in your quality control protocols.
We invite you to collaborate with us to optimize your supply chain for critical impurity standards. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our efficient synthesis route can reduce your overall procurement costs. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. By partnering with NINGBO INNO PHARMCHEM, you gain access to a reliable, high-quality supply of Terbutaline Sulfate Impurity B that supports your commitment to patient safety and regulatory compliance.