Advanced Synthesis of Acemetacin Impurity D for Commercial Pharmaceutical Manufacturing
Advanced Synthesis of Acemetacin Impurity D for Commercial Pharmaceutical Manufacturing
The pharmaceutical industry continuously demands higher purity standards for reference substances to ensure drug safety and regulatory compliance. Patent CN114315687A introduces a groundbreaking methodology for the preparation of Acemetacin Impurity D, a critical related substance for quality control in non-steroidal anti-inflammatory drug (NSAID) manufacturing. This technical disclosure outlines a robust 6-step synthetic pathway starting from the commercially abundant raw material Indomethacin. By shifting the synthetic strategy from traditional ring-building approaches to late-stage functionalization, this innovation addresses long-standing challenges in efficiency and scalability. For global procurement leaders and R&D directors, this represents a significant opportunity to secure high-purity intermediates with improved supply chain resilience. The following analysis details the mechanistic advantages and commercial viability of this novel process.
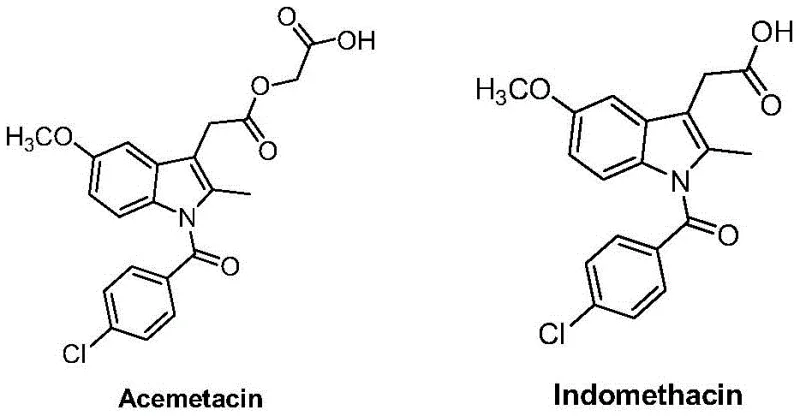
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Acemetacin Impurity D relied on archaic methodologies dating back to the 1980s, which utilized o-tert-butyl phenol as the primary starting material. This conventional route necessitates a cumbersome 12-step linear sequence involving nitration, methyl etherification, nitro reduction, and Fischer indole synthesis to construct the core heterocyclic scaffold. Such extensive synthetic lines inherently accumulate yield losses at every stage, resulting in poor overall atom economy and excessive waste generation. Furthermore, the requirement for specialized phenolic precursors introduces supply chain vulnerabilities and increases raw material costs significantly. The complexity of purifying intermediates at each of the twelve stages further exacerbates production timelines, making this approach unsuitable for modern, cost-sensitive pharmaceutical manufacturing environments where speed and efficiency are paramount.
The Novel Approach
In stark contrast, the patented methodology leverages Indomethacin, a readily available bulk drug substance, as the strategic starting point, effectively bypassing the need for de novo indole ring construction. This innovative route condenses the synthesis into merely 6 steps, focusing on precise functional group manipulation rather than scaffold assembly. By protecting the carboxylic acid moiety early in the sequence, the process enables selective electrophilic aromatic substitution at the 6-position of the indole ring. This strategic simplification not only drastically reduces the consumption of solvents and reagents but also minimizes the formation of hard-to-remove side products. The result is a streamlined workflow that enhances throughput while maintaining rigorous control over the structural integrity of the final impurity standard, offering a superior alternative for industrial-scale production.
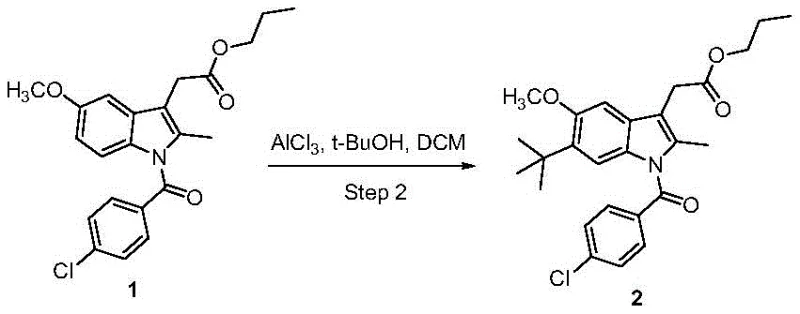
Mechanistic Insights into AlCl3-Catalyzed Friedel-Crafts Alkylation
The cornerstone of this synthetic advancement lies in the regioselective Friedel-Crafts alkylation performed in the second step, where a tert-butyl group is introduced specifically at the 6-position of the indole nucleus. The reaction employs anhydrous aluminum trichloride (AlCl3) as a potent Lewis acid catalyst in conjunction with tert-butanol as the alkylating source. Mechanistically, the Lewis acid activates the tert-butanol to generate a reactive tert-butyl carbocation species, which then attacks the electron-rich indole ring. The presence of the methoxy group at the 5-position and the steric environment created by the protected side chains direct the electrophilic attack exclusively to the 6-position. This high degree of regiocontrol is critical, as substitution at other positions would generate isomeric impurities that are difficult to separate, thereby compromising the purity profile required for analytical reference standards.
Following the alkylation, the process incorporates a strategic hydrolysis and re-functionalization sequence to restore the necessary acetic acid side chain while installing the p-chlorobenzoyl group. The hydrolysis step utilizing lithium hydroxide effectively cleaves the temporary propyl ester and the amide protecting groups, revealing the free indole nitrogen and carboxylic acid. Subsequent O-alkylation with tert-butyl bromoacetate reinstates the acetic acid moiety with orthogonal protection, allowing for the final acylation with p-chlorobenzoyl chloride without interference. This careful orchestration of protection and deprotection strategies ensures that the sensitive indole nitrogen remains available for the final acylation step, ultimately yielding the target Impurity D structure with high fidelity and minimal byproduct formation.
How to Synthesize Acemetacin Impurity D Efficiently
The execution of this synthesis requires precise control over reaction conditions, particularly during the alkylation and hydrolysis phases, to maximize yield and purity. The protocol begins with the O-alkylation of Indomethacin using 1-bromopropane in DMF, followed by the critical Lewis acid-catalyzed tert-butylation. Detailed operational parameters, including stoichiometry, temperature profiles, and workup procedures, are essential for reproducibility. For laboratory and pilot plant teams aiming to implement this route, adherence to the specific molar ratios and solvent volumes described in the patent is crucial to avoid common pitfalls such as over-alkylation or incomplete hydrolysis. The detailed standardized synthesis steps for this process are outlined in the guide below.
- Protect the carboxylic acid of Indomethacin via O-alkylation with 1-bromopropane to form Intermediate 1.
- Perform regioselective Friedel-Crafts alkylation at the 6-position of the indole ring using tert-butanol and AlCl3 to yield Intermediate 2.
- Hydrolyze the ester and amide groups, followed by re-alkylation with tert-butyl bromoacetate and acylation to finalize the structure.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented process offers transformative benefits for procurement managers and supply chain directors seeking to optimize the sourcing of pharmaceutical intermediates. By reducing the synthetic linear steps by half compared to traditional methods, the process inherently lowers the cost of goods sold (COGS) through reduced labor, utility, and material consumption. The reliance on Indomethacin, a commodity chemical with a stable global supply, mitigates the risks associated with sourcing exotic or custom-synthesized starting materials. This shift enhances supply chain reliability, ensuring consistent availability of Acemetacin Impurity D for quality control laboratories and regulatory submissions without the delays often caused by complex multi-step syntheses.
- Cost Reduction in Manufacturing: The elimination of six synthetic steps directly translates to substantial operational savings, as fewer unit operations mean reduced energy consumption and solvent waste disposal costs. The use of common reagents like aluminum trichloride and tert-butanol, rather than expensive specialized catalysts, further drives down the raw material expenditure. Additionally, the simplified purification requirements reduce the burden on downstream processing equipment, allowing for higher batch throughput and better utilization of manufacturing assets. These cumulative efficiencies result in a significantly more cost-effective production model for high-purity pharmaceutical impurities.
- Enhanced Supply Chain Reliability: Sourcing strategies are greatly improved by the use of Indomethacin, which is produced at a massive scale globally, ensuring that raw material shortages are unlikely to disrupt production schedules. The robustness of the reaction conditions, which tolerate standard industrial equipment and do not require cryogenic temperatures or ultra-high vacuum, facilitates manufacturing across diverse geographic locations. This flexibility allows for diversified sourcing strategies, reducing dependency on single-region suppliers and enhancing the overall resilience of the pharmaceutical supply network against geopolitical or logistical disruptions.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions that are easily transferable from laboratory glassware to large-scale stainless steel reactors. The reduction in step count inherently lowers the environmental footprint by minimizing the volume of hazardous waste generated per kilogram of product. Furthermore, the avoidance of heavy metal catalysts or toxic reagents simplifies wastewater treatment and aligns with increasingly stringent environmental regulations. This green chemistry approach not only ensures compliance but also appeals to environmentally conscious stakeholders, adding value to the corporate sustainability profile.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Acemetacin Impurity D. These insights are derived directly from the patent specifications and are intended to clarify the advantages of this novel route for technical decision-makers. Understanding these details is vital for evaluating the feasibility of adopting this method for internal production or external sourcing. Comprehensive answers regarding yield optimization, purity specifications, and regulatory alignment are provided below.
Q: Why is Indomethacin preferred over o-tert-butyl phenol for synthesizing Acemetacin Impurity D?
A: Using Indomethacin as the starting material significantly reduces the linear synthetic steps from 12 to 6, leveraging the existing indole scaffold and avoiding complex early-stage ring construction.
Q: How is regioselectivity controlled during the tert-butyl introduction?
A: The process utilizes a specific Lewis acid catalyst system (anhydrous AlCl3) with tert-butanol under controlled temperatures to ensure exclusive substitution at the 6-position of the indole ring.
Q: What purity levels can be achieved with this novel method?
A: The optimized protocol, including column chromatography purification, consistently delivers Acemetacin Impurity D with purity exceeding 98%, meeting stringent pharmacopoeial standards for reference substances.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Acemetacin Impurity D Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality impurity standards in ensuring drug safety and regulatory compliance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Acemetacin Impurity D meets or exceeds the 98% purity threshold required for analytical applications. Our commitment to technical excellence ensures that you receive a product that supports accurate quantification and robust method validation.
We invite you to collaborate with us to leverage this advanced synthetic technology for your supply chain needs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized manufacturing process can enhance your operational efficiency and reduce total procurement costs.