Advanced Synthesis of 5,6-Hydroxyl Substituted Brazilin Analog for Oncology Applications
Introduction to Novel Brazilin Analog Synthesis
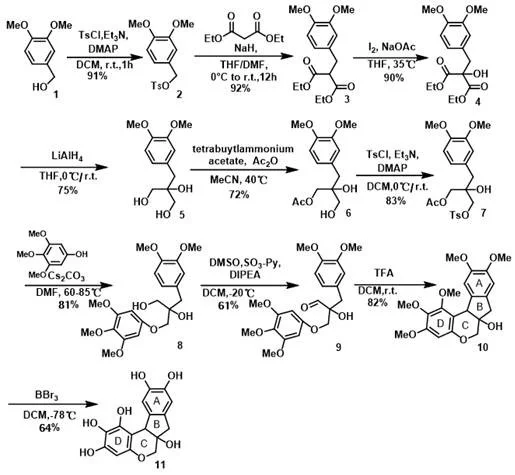
The pharmaceutical industry is constantly seeking efficient pathways to access complex natural product analogs with enhanced therapeutic profiles. Patent CN115181086A discloses a groundbreaking preparation method for a 5,6-position hydroxyl substituted brazilin analogue, a novel compound with significant potential in oncology. This specific analog, characterized by the introduction of hydroxyl groups at the C-5 and C-6 positions of the D ring, represents a structural evolution of traditional brazilin natural products found in sappan wood. The disclosed synthesis strategy leverages a sophisticated sequence of ten chemical transformations, starting from the readily available 3,4-dimethoxybenzyl alcohol. By integrating key reactions such as top Ts protection, SN2 alkylation, iodine-mediated functionalization, and a pivotal Prins/Friedel-Crafts one-pot cyclization, this method achieves a level of efficiency previously unattainable. For R&D directors and procurement specialists, this patent offers a viable alternative to legacy syntheses, promising reduced operational burdens and improved supply chain stability for high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the total synthesis of brazilin and its analogs has been fraught with significant challenges that hinder commercial viability. Prior art, including seminal works by Morsingh (1970), Xu (1996), and Lee (2008), often relies on harsh reaction conditions that demand rigorous control parameters, increasing the risk of batch failure. For instance, gold-catalyzed deoxygenative Nazarov cyclization, while elegant, introduces expensive transition metals that require complex removal processes to meet stringent purity specifications for API manufacturing. Furthermore, traditional routes frequently suffer from low atom economy and the generation of numerous reaction byproducts, necessitating extensive purification steps that erode overall yield. These inefficiencies translate directly into higher production costs and extended lead times, creating bottlenecks for reliable pharmaceutical intermediate suppliers aiming to meet global demand for anti-cancer agents.
The Novel Approach
In stark contrast, the methodology described in CN115181086A presents a streamlined, robust pathway designed for practical scalability. The novel approach utilizes a linear sequence that strategically builds molecular complexity through reliable transformations such as lithium aluminum hydride reduction and Parikh-Doering oxidation. A standout feature of this route is the implementation of a Prins/Friedel-Crafts one-pot reaction to construct the core chroman ring system, which drastically simplifies the synthetic logic compared to multi-step cyclizations found in older literature. The process operates under relatively mild conditions, avoiding extreme temperatures or pressures in most steps, thereby enhancing safety and reproducibility. By minimizing the number of isolation steps and utilizing common chemical reagents, this method effectively reduces synthesis cost and environmental impact, positioning it as a superior choice for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Prins/Friedel-Crafts Cyclization
The cornerstone of this synthetic achievement is the strategic application of the Prins/Friedel-Crafts cascade reaction during the conversion of compound 9 to compound 10. In this critical step, the aldehyde functionality generated via Parikh-Doering oxidation serves as an electrophile that undergoes acid-catalyzed activation by trifluoroacetic acid (TFA) in dichloromethane. This activation triggers an intramolecular cyclization where the electron-rich aromatic ring attacks the activated carbonyl species, forming the essential C-C bonds of the tetracyclic framework. The mechanism proceeds through a stabilized carbocation intermediate, which is subsequently trapped to establish the stereochemistry at the ring junction. This one-pot transformation is particularly advantageous because it combines bond formation and functional group manipulation in a single operation, minimizing the exposure of sensitive intermediates to degradation. For process chemists, understanding this mechanistic nuance is vital for optimizing reaction parameters to maximize the 82% yield reported in the patent examples.
Furthermore, the final demethylation step utilizing boron tribromide (BBr3) at cryogenic temperatures (-78°C) demonstrates precise control over chemoselectivity. This low-temperature condition is crucial for cleaving the methyl ethers at the 5 and 6 positions without compromising the integrity of the rest of the sensitive polyphenolic structure. The use of BBr3 ensures a clean conversion to the target 5,6-dihydroxy substituted brazilin analog (Formula 11) with minimal formation of over-reacted byproducts or decomposition materials. Such meticulous control over the final deprotection stage is essential for achieving the high purity required for biological testing and subsequent drug development. The ability to execute this transformation with a 64% yield underscores the robustness of the overall route, providing a reliable foundation for commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize 5,6-Dihydroxy Brazilin Analog Efficiently
The synthesis of this high-value oncology intermediate begins with the sulfonylation of 3,4-dimethoxybenzyl alcohol, setting the stage for subsequent carbon-carbon bond forming events. The process flows through a series of well-defined steps including alkylation with diethyl malonate, iodine-mediated hydroxylation, and reduction, culminating in the critical cyclization and demethylation phases. Each step has been optimized to balance yield and operational simplicity, making it accessible for laboratories equipped with standard synthetic capabilities. The detailed standardized synthesis steps see the guide below for specific reaction conditions and workup procedures.
- Perform Ts protection on 3,4-dimethoxybenzyl alcohol followed by SN2 reaction with diethyl malonate and iodine-mediated hydroxylation.
- Execute lithium aluminum hydride reduction, acetylation, and subsequent Ts protection to prepare the precursor for ether formation.
- Conduct epoxidation, Parikh-Doering oxidation, and a critical Prins/Friedel-Crafts one-pot cyclization, finishing with low-temperature demethylation.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route offers tangible strategic benefits beyond mere technical elegance. The shift away from precious metal catalysts and exotic reagents towards commodity chemicals like p-toluenesulfonyl chloride, sodium hydride, and acetic anhydride significantly mitigates supply risk. This reliance on widely available raw materials ensures that production schedules are not held hostage by the volatility of specialized reagent markets, thereby enhancing supply chain reliability. Moreover, the simplified purification protocols implied by the reduced byproduct profile mean that manufacturing cycles can be completed faster, effectively reducing lead time for high-purity pharmaceutical intermediates. This efficiency is critical for maintaining continuous supply lines to downstream API manufacturers who operate on tight timelines.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts, such as gold used in previous methodologies, removes the need for costly metal scavenging and validation steps. This fundamental change in the catalyst regime leads to substantial cost savings in raw material expenditure and waste disposal. Additionally, the high yields observed in key steps, such as the 92% yield in the alkylation phase and 90% in the iodination phase, contribute to a superior overall mass balance. By maximizing the conversion of starting materials into the final product, the process minimizes waste generation and lowers the cost per kilogram of the active intermediate, driving significant economic value for large-scale production.
- Enhanced Supply Chain Reliability: The starting material, 3,4-dimethoxybenzyl alcohol, is a commercially abundant chemical, ensuring a stable upstream supply base. Unlike routes dependent on chiral pool materials that may face seasonal or geopolitical shortages, this synthetic approach relies on petrochemical-derived feedstocks with established global logistics networks. The robustness of the reaction conditions, which tolerate minor variations in temperature and stoichiometry without catastrophic failure, further insulates the supply chain from operational disruptions. This resilience allows suppliers to offer more consistent delivery windows and maintain safety stock levels with greater confidence, securing the continuity of supply for critical oncology drug development programs.
- Scalability and Environmental Compliance: The process design inherently supports green chemistry principles by reducing the number of synthetic steps and avoiding toxic heavy metals. The use of standard solvents like dichloromethane, tetrahydrofuran, and ethyl acetate facilitates straightforward solvent recovery and recycling systems, aligning with modern environmental compliance standards. The absence of complex chromatographic separations in favor of crystallization or simple extraction in several steps indicates a route that is amenable to kilogram-to-ton scale manufacturing. This scalability ensures that as clinical demand grows, the production capacity can be expanded seamlessly without requiring entirely new process technologies, supporting the long-term commercial viability of the brazilin analog as a therapeutic agent.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this 5,6-hydroxyl substituted brazilin analogue. These insights are derived directly from the experimental data and claims within the patent documentation, providing clarity on the feasibility and performance of the described technology. Understanding these details is essential for stakeholders evaluating the integration of this intermediate into their drug discovery pipelines.
Q: What is the primary advantage of this new brazilin analog synthesis route?
A: The primary advantage lies in the significantly shortened synthetic steps and mild reaction conditions compared to prior art methods like gold-catalyzed cyclization. This route utilizes conventional reagents such as TsCl and BBr3, reducing operational complexity and byproduct formation while maintaining high stereocontrol.
Q: What is the biological activity of the 5,6-dihydroxy brazilin analog?
A: According to the patent data, the synthesized analog demonstrates potent anti-tumor activity specifically against bladder cancer cell line T24. Experimental results indicate an IC50 value of 31.66 μg/ml after 24 hours of exposure, with inhibition rates reaching over 94% at higher concentrations.
Q: Is this synthesis route scalable for commercial production?
A: Yes, the route is highly scalable as it avoids exotic catalysts and relies on standard unit operations like extraction, distillation, and column chromatography. The use of stable intermediates and robust reaction conditions, such as the Prins/Friedel-Crafts cyclization at room temperature, facilitates safe scale-up from laboratory to industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5,6-Dihydroxy Brazilin Analog Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this novel brazilin analog in the fight against bladder cancer. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to market-ready supply. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of intermediate delivered meets the highest quality standards required for pharmaceutical applications. We are committed to supporting your R&D goals with a supply chain that prioritizes consistency, quality, and regulatory compliance.
We invite you to collaborate with us to leverage this advanced synthetic technology for your oncology portfolio. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this route can optimize your budget. Please contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments. Let us help you accelerate your drug development timeline with a reliable supply of this high-purity pharmaceutical intermediate.