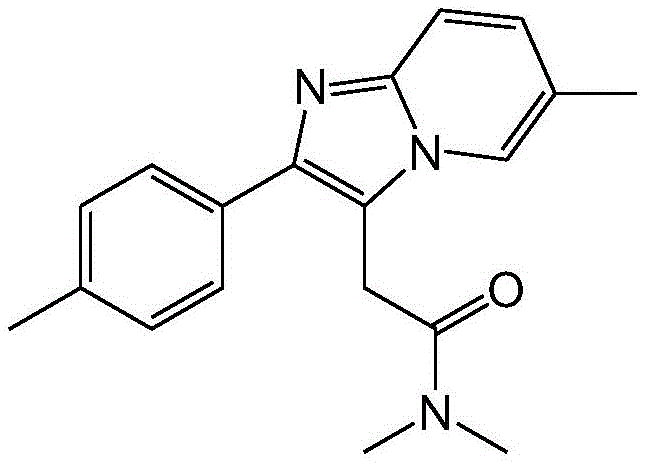
Revolutionizing Zolpidem Production: A One-Step Catalytic Cyclization Strategy for Industrial Scale-Up
Introduction to Advanced Zolpidem Manufacturing Technologies
The pharmaceutical landscape for sedative-hypnotics is constantly evolving, driven by the need for safer, more efficient, and cost-effective manufacturing processes. Patent CN114773337A introduces a groundbreaking methodology for the preparation of zolpidem, a critical active pharmaceutical ingredient (API) widely used for the short-term treatment of insomnia. This invention marks a significant departure from legacy synthetic routes by employing a direct, one-step cyclization reaction between unsaturated ketone compounds and 2-amino-5-methylpyridine. Unlike conventional multi-step sequences that often suffer from low yields and hazardous reagent usage, this novel approach leverages advanced catalytic systems to construct the imidazopyridine core with exceptional efficiency. For R&D directors and procurement specialists, this technology represents a pivotal opportunity to optimize supply chains, reduce reliance on toxic halogenating agents, and achieve higher purity profiles essential for regulatory compliance in global markets.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
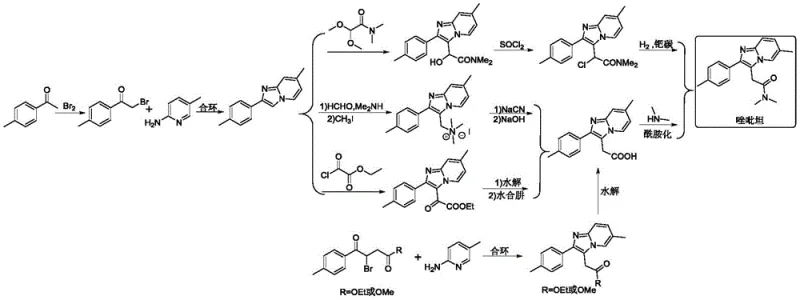
Historically, the industrial synthesis of zolpidem has been plagued by significant technical and safety challenges inherent to older methodologies. The most prevalent traditional route involves the use of alpha-halogenated acetophenones, typically alpha-bromoacetophenone, which are reacted with 2-amino-5-methylpyridine to construct the imidazopyridine ring. This approach necessitates the use of elemental bromine, a highly corrosive substance that demands specialized, expensive equipment and rigorous safety protocols to prevent equipment degradation and personnel exposure. Furthermore, the bromination step inevitably leads to the formation of brominated aromatic hydrocarbon byproducts, which are classified as genotoxic impurities. Removing these trace contaminants requires extensive and costly purification steps, complicating the quality control process and increasing the overall production timeline. Additionally, alternative methods involving palladium-catalyzed coupling or nickel-catalyzed reactions introduce heavy metal residues that must be meticulously scrubbed to meet strict pharmacopeial limits, further driving up operational expenditures and environmental waste.
The Novel Approach
In stark contrast to these cumbersome legacy processes, the method disclosed in CN114773337A offers a streamlined, atom-economical pathway that bypasses the need for halogenation entirely. The core innovation lies in the direct cyclization of an unsaturated ketone precursor (Compound B1) with 2-amino-5-methylpyridine in the presence of a specialized catalyst system. This reaction effectively merges the ring construction and side-chain installation into a single, cohesive operation, drastically reducing the number of unit operations required. By eliminating the bromination step, the process inherently avoids the generation of genotoxic brominated impurities, thereby simplifying the purification workflow and enhancing the safety profile of the final API. The versatility of this method allows for the use of various R1 groups on the ketone precursor, enabling the direct synthesis of zolpidem or its precursors (such as esters or acids) which can be subsequently converted with high efficiency. This strategic simplification not only accelerates the manufacturing cycle but also aligns perfectly with modern green chemistry principles by minimizing waste and hazardous reagent consumption.
Mechanistic Insights into Lewis Acid-Catalyzed Cyclization
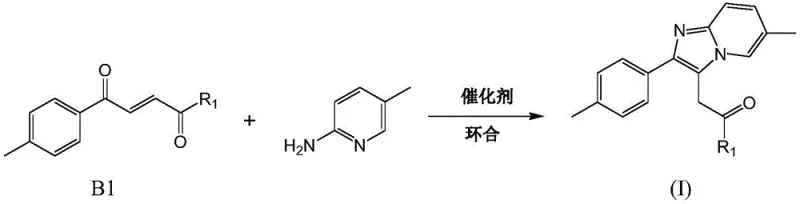
The success of this novel synthetic route hinges on a sophisticated catalytic mechanism that facilitates the nucleophilic attack and subsequent ring closure under mild conditions. The reaction employs a dual-function catalyst system comprising activated molecular sieves (specifically 3A or 4A types) and Lewis acids such as boron trifluoride diethyl etherate, anhydrous cuprous chloride, or phosphorus oxychloride. The molecular sieves play a critical thermodynamic role by adsorbing the water molecules generated during the condensation reaction, thereby shifting the chemical equilibrium towards the product side according to Le Chatelier's principle. Simultaneously, the Lewis acid catalyst activates the carbonyl group of the unsaturated ketone, increasing its electrophilicity and making it more susceptible to nucleophilic attack by the amino group of the pyridine derivative. This activation lowers the energy barrier for the cyclization step, allowing the reaction to proceed efficiently at temperatures ranging from 50°C to 140°C. The choice of solvent, such as chlorobenzene or acetonitrile, further optimizes the solubility of reactants and the stability of the transition states, ensuring a robust and reproducible reaction profile suitable for large-scale batch processing.
From an impurity control perspective, this mechanistic pathway offers distinct advantages over radical-based halogenation methods. Because the reaction proceeds through a polar, ionic mechanism rather than a free-radical pathway, the formation of non-specific side products is significantly suppressed. The specificity of the Lewis acid coordination ensures that the cyclization occurs regioselectively at the desired position on the pyridine ring, minimizing the formation of structural isomers. Furthermore, the absence of heavy metal catalysts like palladium or nickel eliminates the risk of metal leaching, a common pain point in downstream processing. The resulting crude product typically exhibits high purity levels, often exceeding 99% as demonstrated in the patent examples, which reduces the burden on final crystallization steps. This high level of selectivity is crucial for maintaining a clean impurity profile, ensuring that the final zolpidem product meets the stringent specifications required for clinical applications without the need for complex chromatographic separations.
How to Synthesize Zolpidem Efficiently
Implementing this advanced synthesis strategy requires precise control over reaction parameters to maximize yield and purity. The process begins with the careful selection of the unsaturated ketone starting material, which can be prepared via established aldol condensation methods. The cyclization step is conducted in a reactor equipped with efficient stirring and temperature control, where the catalyst loading and reaction time are optimized based on real-time monitoring (HPLC or TLC). Following the reaction, the workup procedure is remarkably straightforward, involving simple filtration to remove the solid molecular sieves, followed by solvent removal and liquid-liquid extraction. The organic phase is then washed with aqueous base to remove acidic impurities and unreacted starting materials. For those aiming for the highest purity, a final recrystallization from a mixed solvent system (such as DCM-MTBE) yields the pharmaceutical-grade product. Detailed standardized operating procedures for each stage of this synthesis, including specific molar ratios and quenching protocols, are outlined in the comprehensive guide below.
- Cyclize unsaturated ketone compound B1 with 2-amino-5-methylpyridine using molecular sieves and a Lewis acid catalyst (e.g., BF3·Et2O) at 50-140°C to form the imidazopyridine core.
- If an ester precursor is formed, perform hydrolysis using inorganic bases to obtain zolpidic acid, followed by amidation with dimethylamine.
- Purify the crude product through solvent extraction, pH adjustment, and recrystallization to achieve high-purity zolpidem suitable for pharmaceutical applications.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this catalytic cyclization technology translates into tangible strategic benefits that extend beyond mere chemical efficiency. The primary advantage lies in the drastic simplification of the raw material portfolio. By replacing hazardous alpha-bromoacetophenones and expensive noble metal catalysts with readily available unsaturated ketones and common Lewis acids, manufacturers can significantly mitigate supply chain risks associated with regulated or scarce reagents. This shift not only stabilizes the sourcing of key inputs but also insulates production costs from the volatility often seen in the markets for bromine and palladium. Furthermore, the reduction in process steps—from multi-step sequences to a direct one-pot cyclization—directly correlates with reduced labor hours, lower energy consumption, and decreased equipment occupancy time. These operational efficiencies cumulatively drive down the cost of goods sold (COGS), allowing for more competitive pricing in the global generic pharmaceutical market while maintaining healthy profit margins.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and corrosive halogenating agents results in substantial savings on raw material procurement. Additionally, the simplified workup process reduces the volume of solvents and reagents required for purification, leading to lower waste disposal costs and reduced utility consumption. The high yield reported in the patent examples (over 70% in several embodiments) further enhances material throughput, ensuring that every kilogram of input generates maximum output value.
- Enhanced Supply Chain Reliability: By utilizing commodity chemicals like 2-amino-5-methylpyridine and substituted chalcones, the supply chain becomes more resilient to disruptions. These materials are produced by a wide range of chemical suppliers globally, reducing dependency on single-source vendors. The robustness of the reaction conditions, which tolerate a range of temperatures and solvents, also means that production can be easily transferred between different manufacturing sites or scaled up without requiring specialized, corrosion-resistant infrastructure.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated in multi-gram to kilogram scales with consistent results. The avoidance of genotoxic impurities simplifies the regulatory filing process, as there is less need for extensive validation of cleaning procedures to remove trace brominated byproducts. Moreover, the reduced generation of hazardous waste aligns with increasingly strict environmental regulations, positioning the manufacturer as a responsible and sustainable partner for major pharmaceutical clients who prioritize green supply chains.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this new synthesis method is vital for stakeholders evaluating its potential for integration into existing production lines. The following questions address common concerns regarding catalyst recovery, substrate scope, and regulatory implications. These answers are derived directly from the technical disclosures within the patent documentation, providing a factual basis for decision-making. Whether you are assessing the feasibility of toll manufacturing or considering a technology transfer, these insights clarify the operational realities of implementing this catalytic cyclization route for zolpidem production.
Q: How does this new method eliminate genotoxic impurities compared to traditional routes?
A: Traditional methods often rely on alpha-halogenated acetophenones, which introduce brominated aromatic hydrocarbon impurities that are genotoxic. This novel cyclization method avoids halogenation entirely, utilizing a direct condensation reaction that significantly reduces the risk of such hazardous byproducts.
Q: What catalysts are used in the patented zolpidem synthesis process?
A: The process utilizes a combination of activated molecular sieves (3A or 4A) to remove water and drive the equilibrium, alongside Lewis acid catalysts such as boron trifluoride diethyl etherate, anhydrous cuprous chloride, or phosphorus tribromide to facilitate the ring closure.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the method is designed for industrial scalability. It uses readily available raw materials, operates at moderate temperatures (50-140°C), and involves simple workup procedures like filtration and extraction, making it highly adaptable for metric-ton scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Zolpidem Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to advanced synthetic routes requires a partner with deep technical expertise and proven industrial capability. As a leading CDMO specializing in complex pharmaceutical intermediates, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of Lewis acid-catalyzed reactions, including moisture-sensitive operations and precise temperature control. We adhere to stringent purity specifications and operate rigorous QC labs to ensure that every batch of zolpidem or its precursors meets the highest international standards. Our team is ready to assist you in optimizing this novel cyclization process, ensuring a seamless transition from laboratory scale to full commercial manufacturing.
We invite you to engage with our technical procurement team to discuss how this innovative method can enhance your supply chain efficiency. By leveraging our capabilities, you can secure a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage potential partners to request specific COA data and route feasibility assessments to validate the superior quality and economic advantages of our zolpidem production technology. Let us collaborate to build a more resilient and cost-effective supply chain for this critical therapeutic agent.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →