Advanced Stereospecific Synthesis of Picropodophyllin Intermediates for Commercial API Production
The pharmaceutical industry continuously seeks robust synthetic pathways for complex antineoplastic agents, particularly those derived from the podophyllotoxin family. Patent CN1021822C discloses a novel, highly efficient, and stereospecific synthesis of picropodophyllin and its related compounds, which serve as critical precursors for known anticancer drugs such as etoposide and teniposide. Unlike traditional extraction methods from natural sources which are limited by supply variability, this total synthesis approach offers a reliable pharmaceutical intermediate supplier solution by establishing a consistent, scalable chemical route. The invention specifically addresses the challenges associated with controlling the stereochemistry at the C-4 position, distinguishing the 4-hydroxy epimer (picropodophyllin) from the naturally occurring podophyllotoxin. By leveraging advanced cycloaddition chemistry, this technology provides a strategic advantage for manufacturers aiming to secure high-purity intermediates for oncology drug production.
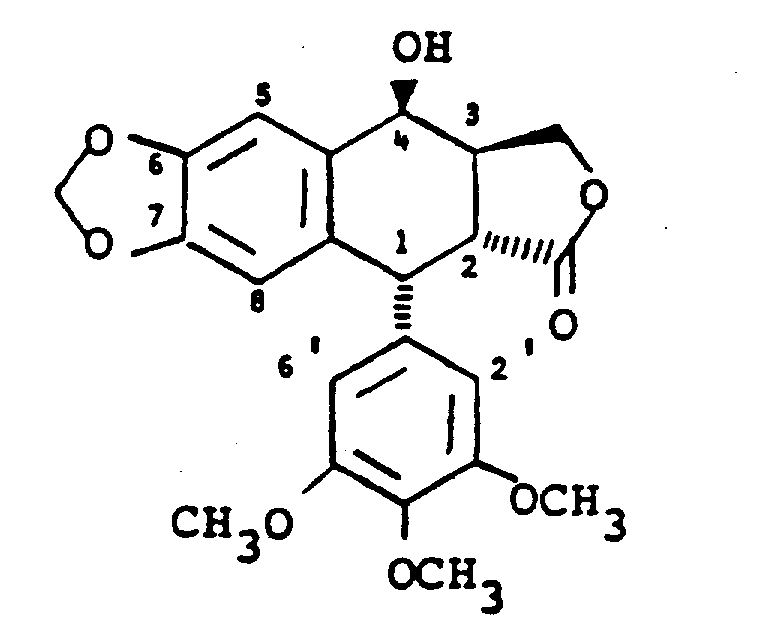
The development of this methodology was driven by the significant limitations inherent in conventional synthetic routes previously described in literature, such as those by Gensler, Kende, and Murphy. Traditional approaches often relied on the epimerization of adjacent tetrahydropyran derivatives or lengthy multi-step sequences totaling 12 to 13 steps with disappointingly low overall yields, sometimes as low as 4.5%. A major bottleneck in these legacy processes was the thermodynamic equilibration during epimerization, which frequently resulted in inseparable mixtures of podophyllotoxin and picropodophyllin in ratios around 45:55, necessitating costly and inefficient purification steps. Furthermore, earlier methods required the preparation of specific dicyclization precursors to achieve satisfactory yields, adding complexity and cost to the manufacturing process. These inefficiencies posed substantial barriers to cost reduction in API manufacturing, making the final antineoplastic agents expensive and difficult to source in bulk quantities.
In stark contrast, the novel approach detailed in this patent circumvents these historical hurdles by introducing a streamlined pathway that avoids the preparation of the picropodophyllin intermediate entirely until the final stages, focusing instead on versatile aryl tetralone precursors. The core innovation lies in the utilization of a [3+2] cycloaddition reaction between a cis-alkene and a substituted nitrile oxide, which constructs the necessary heterocyclic framework with exceptional stereocontrol. This method allows for the direct formation of isoxazole adducts that can be selectively manipulated to reveal the desired functionality without scrambling the stereocenters. Additionally, the patent describes an improved cyclization process for preparing aryl tetralones from cyclopropyl ketones via enol acetates, utilizing Lewis acids and acid anhydrides to achieve high yields rapidly. This represents a paradigm shift in synthetic efficiency, offering a viable route for the commercial scale-up of complex pharmaceutical intermediates.
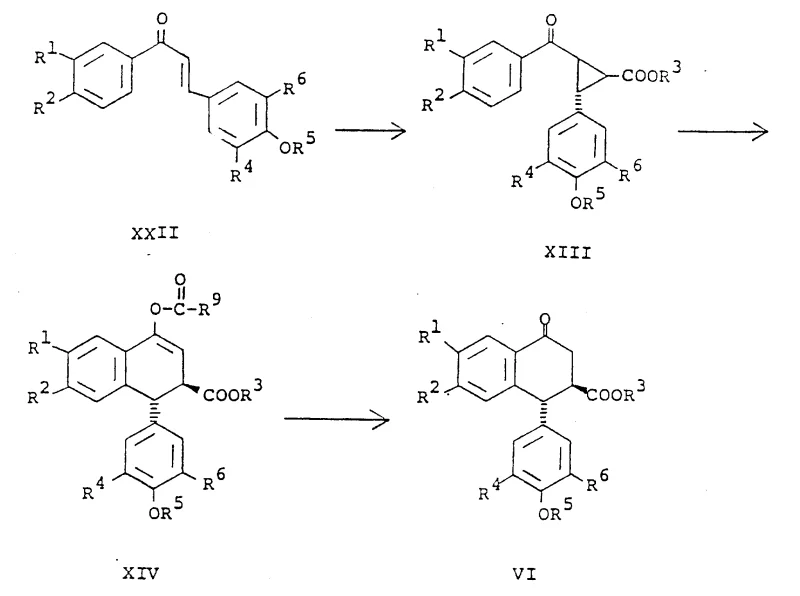
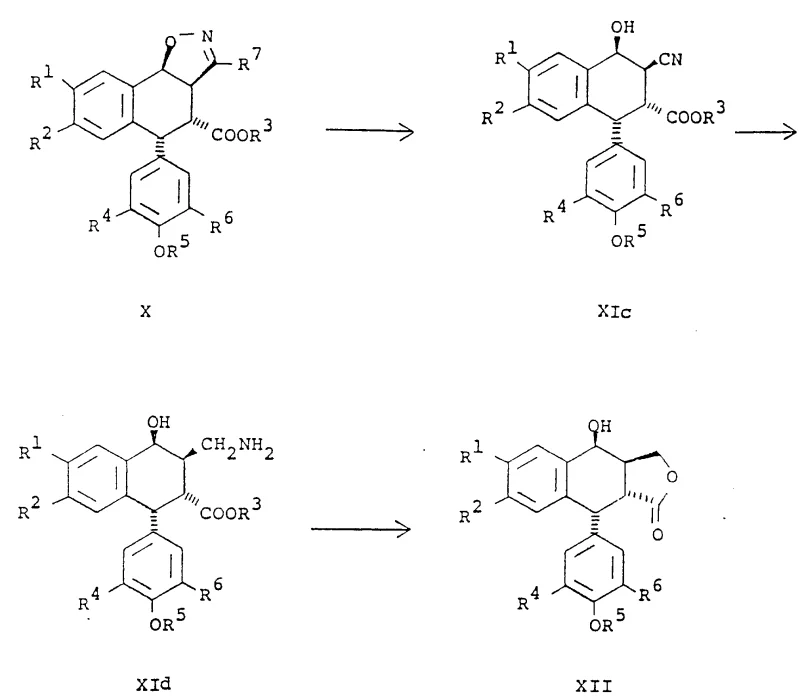
Mechanistically, the success of this synthesis hinges on the precise control of the [3+2] dipolar cycloaddition and the subsequent reductive cleavage of the isoxazole ring. The reaction between the cis-alkene (Formula IX) and the nitrile oxide (Formula XX) proceeds with high regiospecificity, attaching the isoxazole ring to the beta-face of the alkene system. The choice of substituent on the nitrile oxide (R7), such as bromine, chlorine, or cyano groups, is critical as it dictates the conditions required for ring opening. For instance, bromo-isoxazoles can be reduced using radical initiators like tributyltin hydride or catalytic hydrogenation with Raney Nickel, while cyano-substituted variants allow for thermal rearrangement or hydrolysis pathways. This mechanistic flexibility ensures that impurities arising from non-selective addition are minimized, as the steric environment of the cis-alkene directs the incoming dipole to the correct face. The subsequent transformation of the nitrile group into an aminomethyl function, followed by diazotization-induced lactonization, locks the stereochemistry into the final cis-lactone configuration found in picropodophyllin.
For research and development teams looking to implement this technology, the process offers a clear and reproducible workflow for synthesizing these high-value intermediates efficiently. The synthesis begins with the preparation of the cis-alkene precursor, followed by the crucial cycloaddition step with a generated nitrile oxide species in an inert solvent. Detailed experimental protocols within the patent describe specific conditions, such as using nitromethane or ethyl acetate as solvents and maintaining temperatures ranging from -20°C to reflux depending on the specific nitrile oxide substituent. The downstream processing involves selective reduction steps that preserve the sensitive ester and ether functionalities present in the molecule. To facilitate the practical adoption of this chemistry, the detailed standardized synthesis steps see the guide below which outlines the critical operational parameters for maximizing yield and purity.
From a procurement and supply chain perspective, the adoption of this synthetic route offers compelling commercial advantages that address common pain points in the sourcing of oncology intermediates. First and foremost, the streamlined nature of the synthesis contributes to significant cost savings in manufacturing by eliminating the need for extensive chromatographic purifications associated with separating epimeric mixtures in older methods. The ability to use readily available starting materials, such as substituted phenyl styryl ketones which can be prepared from common aromatic aldehydes and ketones, enhances supply chain reliability and reduces dependency on scarce natural extracts. Furthermore, the robustness of the Lewis acid-catalyzed cyclization steps allows for easier process optimization and scale-up, ensuring consistent batch-to-batch quality. The environmental compliance aspect is also improved, as the process avoids the use of certain heavy metal catalysts in favor of more manageable reagents, simplifying waste treatment protocols and aligning with modern green chemistry initiatives in the fine chemical sector.
Frequently Asked Questions regarding this technology often center on its applicability to various derivatives and its regulatory standing. The patent explicitly covers a broad range of substituents (R1 through R9), indicating that the chemistry is adaptable for synthesizing various analogues beyond just the parent picropodophyllin structure, which is valuable for medicinal chemistry campaigns seeking new drug candidates. Additionally, the stereospecific nature of the route means that the resulting intermediates possess a defined chirality, reducing the regulatory burden associated with characterizing racemic mixtures or unknown impurities. Understanding these technical nuances is essential for stakeholders evaluating the feasibility of integrating this pathway into their existing production portfolios for antineoplastic agents.
- Perform a stereoselective [3+2] cycloaddition reaction between a cis-alkene intermediate (Formula IX) and a substituted nitrile oxide (Formula XX) in an inert solvent to generate an isoxazole adduct (Formula X).
- Execute the heterolytic fission of the N-O bond in the isoxazole ring under selective reduction conditions (e.g., using Raney Nickel or tributyltin hydride) to produce a hydroxynitrile intermediate (Formula XIc).
- Reduce the nitrile group to an aminomethyl group (Formula XId) and subsequently induce cyclization via diazotization in acidic media to form the final lactone structure (Formula XII).
Frequently Asked Questions (FAQ)
Q: How does this synthesis method improve upon conventional Gensler or Kende routes?
A: Conventional methods often require 12 to 13 steps with low overall yields (around 4.5%) and involve difficult epimerization steps that produce mixtures of podophyllotoxin and picropodophyllin. This novel process utilizes a highly stereoselective [3+2] cycloaddition that avoids thermodynamic equilibration issues, significantly reducing the step count and improving the purity of the desired cis-configured intermediates.
Q: What represents the key stereochemical control point in this process?
A: The critical stereocontrol is achieved during the [3+2] cycloaddition of the nitrile oxide to the cis-alkene. The reaction proceeds with high regiospecificity and stereoselectivity on the beta-face of the cis-alkene, ensuring the correct spatial arrangement of substituents required for the final lactone ring closure without needing complex separation of diastereomers.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process is designed for commercial feasibility. It utilizes readily available starting materials such as phenyl styryl ketones and avoids the use of expensive or hazardous reagents where possible. The elimination of difficult chromatographic separations associated with older epimerization methods enhances the scalability and supply chain reliability for producing antineoplastic agents like etoposide.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Picropodophyllin Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing high-quality intermediates for the production of life-saving antineoplastic medications. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in patent CN1021822C can be translated into industrial reality. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of picropodophyllin intermediate meets the exacting standards required for pharmaceutical grade API synthesis. Our commitment to technical excellence allows us to navigate the complexities of stereoselective synthesis, delivering products that support your drug development timelines effectively.
We invite potential partners to engage with our technical procurement team to discuss how this advanced synthesis route can be tailored to your specific volume and quality requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how implementing this novel pathway might optimize your overall production costs. We encourage you to contact us directly to obtain specific COA data and route feasibility assessments, ensuring that your supply chain for these critical cancer treatment intermediates remains robust, compliant, and economically viable in the long term.