Advanced Synthesis of Aromatic Aminoalcohol Intermediates for Antidiabetic Drug Manufacturing
Advanced Synthesis of Aromatic Aminoalcohol Intermediates for Antidiabetic Drug Manufacturing
The pharmaceutical landscape for metabolic disorders continues to evolve, driven by the demand for safer and more effective antidiabetic and antiobesity agents. Patent CN1033750C discloses a series of novel intermediate compounds specifically designed for the preparation of aromatic aminoalcohol derivatives featuring a 2-[2-(substituted phenoxy)-1-methylethyl]aminoethanol structure. These intermediates are not merely academic curiosities; they represent a strategic breakthrough in the synthesis of thiazolidinedione-based drugs, which are known for their ability to suppress aldose reductase and manage hyperglycemia. For R&D directors and procurement specialists, understanding the chemical architecture of these intermediates is crucial. The patent outlines a sophisticated approach to constructing the core pharmacophore, utilizing specific protecting groups and stereoselective reactions to ensure the final API meets stringent regulatory standards. By leveraging these novel pathways, manufacturers can achieve superior control over impurity profiles, a critical factor in the commercial viability of any new antidiabetic candidate.
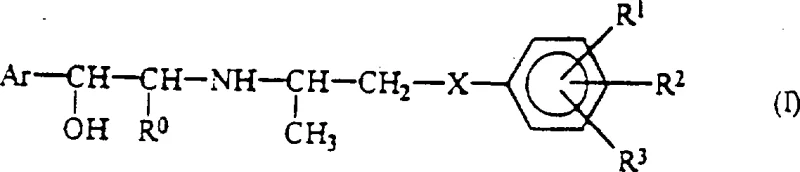
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for aromatic aminoalcohol derivatives often suffer from significant drawbacks that hinder large-scale production. Conventional methods frequently rely on non-selective alkylation reactions that generate complex mixtures of regioisomers and stereoisomers, necessitating expensive and time-consuming purification steps such as preparative HPLC. Furthermore, the thiazolidinedione ring, a key structural motif in this class of drugs, is susceptible to degradation under harsh acidic or basic conditions often employed in older synthesis protocols. This instability can lead to ring-opening byproducts that are difficult to remove and may pose toxicological risks. Additionally, achieving high enantiomeric excess in the side chain without using costly chiral catalysts or resolution agents has historically been a bottleneck, resulting in low overall yields and inflated manufacturing costs. These inefficiencies create substantial supply chain vulnerabilities, making it difficult to secure reliable sources of high-purity intermediates for clinical and commercial needs.
The Novel Approach
The methodology presented in CN1033750C offers a transformative solution to these historical challenges by introducing a modular and highly controlled synthetic strategy. The core innovation lies in the use of specific intermediate compounds, such as 5-(4-acetoxybenzyl)-3-trityl-thiazolidine-2,4-dione, which serve as stable platforms for subsequent functionalization. By employing a trityl protecting group on the nitrogen atom of the thiazolidinedione ring, the process effectively masks the nucleophilic site, preventing unwanted side reactions during the coupling with the phenoxy-alkyl chain. This strategic protection allows for milder reaction conditions that preserve the integrity of the sensitive heterocyclic core. Moreover, the patent details a reductive amination pathway that facilitates the introduction of chiral amines with high stereocontrol, bypassing the need for difficult resolution steps later in the synthesis. This approach not only streamlines the workflow but also significantly enhances the purity of the final product, addressing the critical quality attributes required by modern regulatory agencies.
Mechanistic Insights into Reductive Amination and Protective Group Chemistry
The chemical elegance of this process is best understood through the lens of its mechanistic steps, particularly the reductive amination and the manipulation of hydroxyl-protecting groups. The synthesis begins with the condensation of an amine intermediate, such as formula (XI), with a carbonyl compound of formula (XIII) or (XVI). This reaction forms an imine or iminium ion intermediate, which is subsequently reduced in situ using mild reducing agents like sodium cyanoborohydride or sodium triacetoxyborohydride. The choice of reducing agent is critical; it must be selective enough to reduce the imine without affecting other reducible functionalities present in the molecule, such as the ester or ether linkages. The patent emphasizes the importance of solvent selection, recommending polar aprotic solvents like DMF or alcoholic solvents like methanol to facilitate the reaction kinetics while maintaining solubility of the polar intermediates. This careful balancing of reagents ensures that the coupling efficiency remains high, minimizing the formation of secondary amines or over-reduced byproducts.
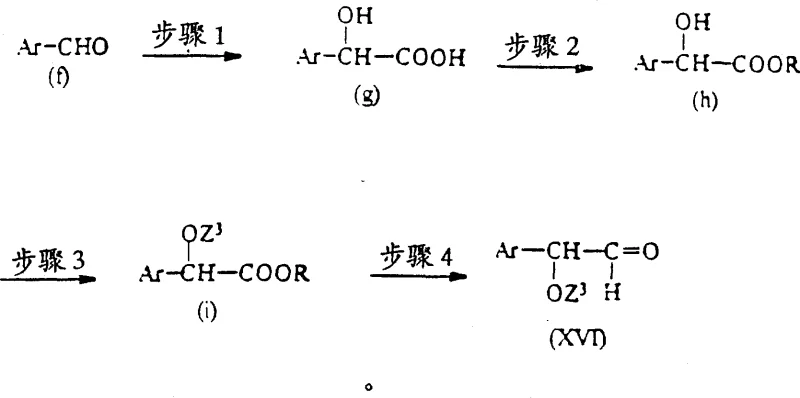
Furthermore, the management of stereochemistry and functional group tolerance is achieved through the judicious use of protecting groups denoted as Z3 in the patent formulas. Groups such as tert-butyldimethylsilyl (TBDMS), tetrahydropyranyl (THP), or trityl are employed to mask hydroxyl functionalities during the construction of the carbon backbone. These groups are orthogonal, meaning they can be removed selectively under specific conditions without disturbing the rest of the molecule. For instance, silyl ethers can be cleaved using fluoride sources like tetrabutylammonium fluoride, while trityl groups are acid-labile. This orthogonality provides chemists with the flexibility to build complex molecular architectures step-by-step. The patent also highlights the preparation of chiral aldehydes via the reduction of esters using diisobutylaluminum hydride (DIBAL-H) at low temperatures. This step is pivotal for establishing the correct stereochemistry at the benzylic position, which is often essential for the biological activity of the final antidiabetic agent. By controlling these mechanistic variables, the process ensures a robust and reproducible synthesis suitable for industrial application.
How to Synthesize Aromatic Aminoalcohol Intermediates Efficiently
Implementing this synthesis requires a precise understanding of the reaction parameters outlined in the patent to ensure optimal yield and purity. The process generally involves the initial preparation of the thiazolidinedione core, followed by protection, coupling, and final deprotection. Each step must be monitored closely to prevent the accumulation of impurities that could complicate downstream processing. The use of catalytic hydrogenation for the reduction of the benzylidene double bond is a key operational step that offers excellent atom economy and scalability. Detailed standard operating procedures for these transformations are critical for technology transfer from the laboratory to the pilot plant. For a comprehensive breakdown of the specific reaction conditions, stoichiometry, and workup procedures, please refer to the standardized synthesis guide below.
- Condense p-Hydroxybenzaldehyde with thiazolidine-2,4-dione to form the benzylidene intermediate, followed by catalytic hydrogenation.
- Protect the nitrogen atom of the thiazolidinedione ring using a trityl group to prevent side reactions during subsequent coupling.
- Perform reductive amination between the protected thiazolidinedione amine and a chiral aldehyde intermediate using sodium cyanoborohydride.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the synthetic routes described in CN1033750C offers tangible benefits that extend beyond mere chemical novelty. The primary advantage lies in the significant simplification of the manufacturing process, which directly translates to cost reduction in API manufacturing. By utilizing stable, crystalline intermediates that can be purified via simple recrystallization rather than column chromatography, the process eliminates one of the most expensive and waste-generating steps in fine chemical production. This reduction in processing complexity lowers the cost of goods sold (COGS) and reduces the environmental footprint associated with solvent consumption and waste disposal. Furthermore, the reliance on commodity chemicals such as p-hydroxybenzaldehyde and thiazolidine-2,4-dione as starting materials ensures a secure and resilient supply chain, mitigating the risk of raw material shortages that often plague specialized synthetic routes.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts in certain steps and the use of robust, scalable reactions like hydrogenation significantly lower operational expenses. The process avoids the need for exotic reagents or cryogenic conditions that require specialized equipment, allowing production to take place in standard multipurpose reactors. This accessibility means that manufacturing can be distributed across a wider network of qualified CDMOs, fostering competition and driving down prices. Additionally, the high selectivity of the reductive amination step minimizes the loss of valuable chiral building blocks, ensuring that the maximum amount of raw material is converted into the desired product. These efficiencies compound over large production volumes, resulting in substantial cost savings that can be passed on to the end customer or reinvested in further R&D.
- Enhanced Supply Chain Reliability: The modular nature of this synthesis allows for the stocking of key intermediates, such as the protected thiazolidinedione, which have long shelf lives and stable storage requirements. This capability enables manufacturers to build strategic inventory buffers, ensuring continuity of supply even in the face of market fluctuations or logistical disruptions. The use of common solvents and reagents further enhances supply security, as these materials are readily available from multiple global suppliers, reducing dependency on single-source vendors. By decoupling the synthesis of the core heterocycle from the final coupling step, production schedules can be optimized to match demand, reducing lead times for high-purity pharmaceutical intermediates. This flexibility is invaluable for meeting the rigorous timelines of drug development programs and commercial launches.
- Scalability and Environmental Compliance: The reaction conditions described are inherently scalable, moving seamlessly from gram-scale laboratory experiments to multi-ton commercial production without significant re-optimization. The processes operate at moderate temperatures and pressures, reducing energy consumption and enhancing safety profiles in large-scale reactors. From an environmental perspective, the avoidance of heavy metal catalysts and the minimization of hazardous waste streams align with green chemistry principles and increasingly strict regulatory standards. The ability to recycle solvents and recover byproducts further improves the sustainability metrics of the manufacturing process. This commitment to environmental stewardship not only reduces compliance risks but also enhances the corporate social responsibility profile of the supply chain, a factor that is becoming increasingly important for major pharmaceutical buyers.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these novel intermediates. The answers are derived directly from the technical specifications and experimental data provided in the patent literature, ensuring accuracy and relevance for industry professionals. Understanding these details is essential for evaluating the feasibility of integrating these compounds into your existing drug development pipelines.
Q: What are the critical purity specifications for these thiazolidinedione intermediates?
A: High purity is essential to minimize genotoxic impurities. The patented process utilizes crystallization and selective protection strategies to achieve pharmaceutical-grade purity without extensive chromatography.
Q: Is the synthesis route scalable for commercial API production?
A: Yes, the route relies on robust unit operations such as catalytic hydrogenation and standard reflux conditions, which are easily transferable from pilot plant to multi-ton commercial manufacturing.
Q: How does the protective group strategy impact overall yield?
A: The use of trityl and silyl protecting groups allows for orthogonal deprotection, significantly reducing byproduct formation and improving the overall yield of the final active pharmaceutical ingredient.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aromatic Aminoalcohol Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of antidiabetic therapies. Our team of expert chemists has extensively analyzed the synthetic pathways disclosed in CN1033750C and possesses the technical capability to execute these complex routes with precision. We offer extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can grow seamlessly from clinical trials to full-scale market supply. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of aromatic aminoalcohol derivatives meets the highest international standards. We understand that time-to-market is crucial, and our optimized processes are designed to deliver results without compromising on quality or safety.
We invite you to collaborate with us to leverage these advanced synthetic technologies for your next generation of metabolic drugs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. By partnering with us, you gain access to specific COA data and route feasibility assessments that will empower your decision-making process. Contact us today to discuss how we can support your supply chain with reliable, cost-effective, and high-purity pharmaceutical intermediates.