Advanced Four-Step Synthesis Strategy for Tomivosertib Kinase Inhibitor Manufacturing
Advanced Four-Step Synthesis Strategy for Tomivosertib Kinase Inhibitor Manufacturing
The pharmaceutical industry is constantly seeking more efficient pathways to produce complex kinase inhibitors, particularly for oncology applications where supply chain reliability is paramount. Patent CN114671869B introduces a groundbreaking synthetic methodology for Tomivosertib (EFT-508), a potent and selective MNK1 and MNK2 kinase inhibitor currently in Phase 2 clinical trials for various solid tumors. This novel approach fundamentally restructures the manufacturing landscape by condensing the synthesis into a concise four-step sequence that prioritizes cost-efficiency and operational simplicity. Unlike traditional routes that often suffer from lengthy linear sequences and expensive reagents, this method leverages robust catalytic hydrogenation and one-pot cyclization strategies. The result is a process that not only achieves exceptional chemical purity but also aligns perfectly with the rigorous demands of Good Manufacturing Practice (GMP) for active pharmaceutical ingredients. For global procurement teams, this represents a significant opportunity to secure a stable supply of high-quality intermediates.
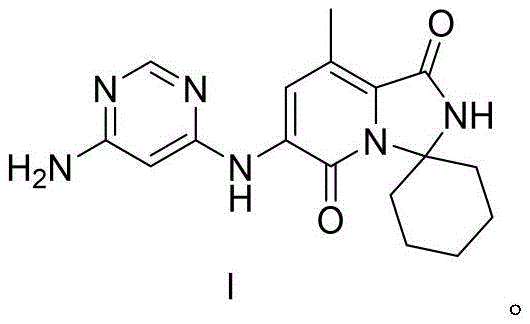
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex heterocyclic kinase inhibitors like Tomivosertib has been plagued by inefficiencies that drive up costs and extend lead times. Conventional routes often rely on multiple protection and deprotection cycles, requiring harsh reagents that generate substantial hazardous waste. Furthermore, traditional methods frequently utilize expensive transition metal catalysts that are difficult to remove to trace levels, posing a significant risk for regulatory compliance in final drug products. The accumulation of impurities across long synthetic sequences often necessitates repetitive chromatographic purifications, which are notoriously difficult to scale beyond kilogram quantities. These bottlenecks create fragility in the supply chain, where a failure in any single step can halt the entire production campaign. Additionally, the reliance on specialized starting materials that are not commodity chemicals exacerbates the risk of supply disruption, making it challenging for manufacturers to guarantee consistent delivery schedules to downstream API producers.
The Novel Approach
The methodology disclosed in patent CN114671869B offers a transformative solution by streamlining the construction of the Tomivosertib core through a highly convergent strategy. As illustrated in the reaction scheme below, the process initiates with a cost-effective reduction using Zinc powder and Palladium on Carbon, followed by a clever one-pot cyclization that constructs the spiro-cyclic framework in a single operation. This eliminates the need for isolating unstable intermediates, thereby reducing material loss and solvent consumption. The subsequent coupling reaction employs a robust Palladium-catalyzed system with Xantphos ligand, ensuring high conversion rates under relatively mild thermal conditions. Finally, the deprotection step utilizes hydrochloric acid in dioxane, a standard industrial reagent that allows for simple workup procedures. This holistic redesign of the synthetic route directly addresses the pain points of scalability and cost, making it an ideal candidate for reliable pharmaceutical intermediate supplier partnerships aiming for commercial scale-up of complex pharmaceutical intermediates.
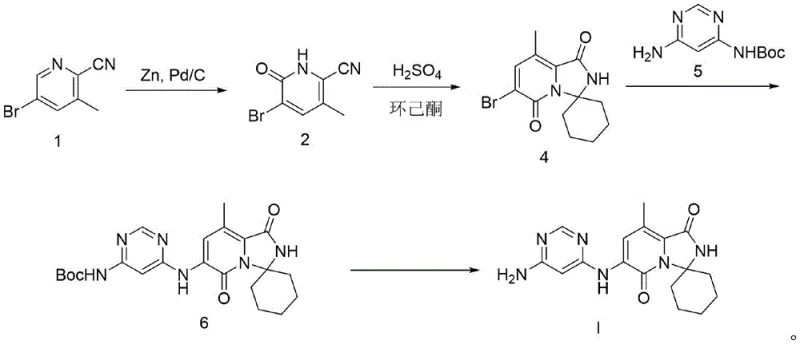
Mechanistic Insights into Pd-Catalyzed Cyclization and Coupling
The heart of this synthetic innovation lies in the precise control of reaction mechanisms to maximize yield and minimize byproduct formation. The initial reduction step converts the nitrile precursor into an amide intermediate using Zinc and Pd/C in water at elevated temperatures between 120°C and 150°C. This heterogeneous catalytic system is advantageous because it avoids the use of pyrophoric reagents like Lithium Aluminum Hydride, significantly enhancing operational safety. The subsequent cyclization involves a nucleophilic addition of the newly formed amide to cyclohexanone, promoted by concentrated sulfuric acid. This acid-catalyzed dehydration drives the equilibrium towards the formation of the spiro-fused ring system, a critical structural motif for the biological activity of the inhibitor. By optimizing the mass ratio of cyclohexanone to the substrate, the process ensures complete conversion while preventing polymerization side reactions. The careful control of pH during the workup, specifically adjusting to a range of 6.8 to 7.1, is crucial for precipitating the product while keeping acidic impurities in the aqueous phase.
Furthermore, the coupling reaction in step three demonstrates sophisticated ligand design to facilitate the formation of the C-N bond between the brominated core and the pyrazine amine. The use of Pd(OAc)2 combined with the bidentate ligand Xantphos creates a highly active catalytic species capable of overcoming the steric hindrance of the substrates. Operating at temperatures around 100°C in 1,4-dioxane under nitrogen protection prevents oxidation of the catalyst and ensures a clean reaction profile. The stoichiometry is finely tuned, with a slight excess of the amine coupling partner to drive the reaction to completion without generating excessive waste. Impurity control is further enhanced by the selectivity of this catalytic system, which minimizes homocoupling of the aryl halide. The final deprotection step is equally critical, where the use of 4M HCl in dioxane at 0°C ensures the rapid cleavage of the Boc group while preserving the integrity of the sensitive amide bonds within the molecule, ultimately delivering the target compound with an HPLC purity of 99.7%.
How to Synthesize Tomivosertib Efficiently
Implementing this synthesis requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and safety. The process begins with the preparation of the amide intermediate, followed immediately by the cyclization to lock in the core structure. Detailed operational guidelines regarding temperature ramps, stirring speeds, and quenching protocols are essential for maintaining the high yields reported in the examples. For R&D teams looking to transfer this technology to pilot plants, understanding the thermodynamics of the exothermic cyclization step is vital for reactor design. The following guide outlines the standardized workflow derived from the patent examples, providing a clear roadmap for laboratory and pilot-scale execution. Please refer to the specific technical documentation for the full Standard Operating Procedures (SOPs).
- Perform catalytic reduction of the brominated precursor using Zinc powder and Pd/C in water at 150°C to form the amide intermediate.
- Execute a one-pot cyclization by reacting the intermediate with concentrated sulfuric acid and cyclohexanone at 100°C.
- Conduct a Buchwald-Hartwig coupling reaction using Pd(OAc)2 and Xantphos ligand to attach the protected pyrazine amine.
- Finalize the synthesis by removing the Boc protecting group using 4M HCl in dioxane at 0°C to yield high-purity Tomivosertib.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route translates into tangible strategic benefits that go beyond simple unit price reductions. The simplification of the process from potentially dozens of steps down to just four major operations drastically reduces the cumulative yield loss typically seen in linear syntheses. This efficiency gain means that less raw material is required to produce the same amount of final product, effectively lowering the cost of goods sold (COGS) without compromising on quality standards. Moreover, the use of commodity chemicals such as Zinc powder, cyclohexanone, and sulfuric acid mitigates the risk associated with sourcing specialized reagents that may have volatile markets or long lead times. This stability in raw material sourcing is a key factor in ensuring business continuity for long-term API manufacturing contracts.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous reagents in favor of robust catalytic systems significantly lowers the operational expenditure. By utilizing a one-pot cyclization strategy, the process reduces solvent usage and energy consumption associated with multiple isolation and drying steps. The high efficiency of the Pd-catalyzed coupling minimizes the loading of precious metals required, and the ease of purification reduces the burden on downstream processing facilities. These factors combine to create a lean manufacturing process that is inherently more cost-competitive in the global market for high-purity pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials ensures that production is not held hostage by the supply constraints of niche chemicals. The robustness of the reaction conditions, such as the tolerance of the catalytic system to minor variations, allows for consistent batch-to-batch quality. This predictability is essential for supply chain planners who need to forecast inventory levels accurately. Furthermore, the simplified workflow reduces the overall cycle time from raw material intake to finished goods, enabling faster response times to fluctuating market demands and reducing the lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process is designed with industrial scale-up in mind, utilizing standard unit operations like filtration, extraction, and distillation that are easily implemented in existing multipurpose plants. The reduction in step count inherently lowers the volume of waste generated per kilogram of product, aligning with green chemistry principles and reducing disposal costs. The use of water in the initial reduction step and the ability to recover solvents like ethyl acetate and dioxane further enhances the environmental profile. This makes the technology not only economically viable but also sustainable, meeting the increasingly stringent environmental regulations faced by modern chemical manufacturers.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Tomivosertib synthesis method. These answers are derived directly from the experimental data and beneficial effects described in the patent literature. They are intended to provide clarity on the feasibility and advantages of adopting this route for commercial production. Understanding these details helps stakeholders make informed decisions about technology transfer and partnership opportunities.
Q: What are the key advantages of this new Tomivosertib synthesis route?
A: The patented method reduces the total synthesis to just four steps, utilizing cheap and readily available raw materials like Zinc powder and cyclohexanone. It features a high-yielding one-pot cyclization and mild deprotection conditions, achieving final HPLC purity of 99.7%.
Q: How does this process improve scalability for industrial production?
A: By eliminating complex multi-step sequences and using robust catalytic systems like Pd/C and Pd(OAc)2/Xantphos, the process minimizes purification difficulties. The use of standard solvents like ethyl acetate and dioxane facilitates easier solvent recovery and waste management on a large scale.
Q: What represents the critical quality control point in this synthesis?
A: The final deprotection step using HCl/dioxane is critical, as controlling the temperature at 0°C and reaction time ensures the removal of the Boc group without degrading the sensitive kinase inhibitor core, guaranteeing the specified 99.7% purity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tomivosertib Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a robust and scalable supply chain for next-generation oncology therapeutics. Our team of expert chemists has thoroughly analyzed the synthetic pathway described in patent CN114671869B and is fully prepared to execute this technology at commercial scales. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from the laboratory to the marketplace. Our state-of-the-art facilities are equipped with the necessary reactors for high-pressure hydrogenation and precise temperature control required for the cyclization and coupling steps. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Tomivosertib intermediate meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to leverage this advanced synthesis method for your pipeline. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. By partnering with us, you gain access to our deep process knowledge and supply chain network. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how we can become your trusted partner in bringing this vital kinase inhibitor to patients worldwide.