Advanced Organocatalytic Synthesis of Xanthone-Spliced Oxindole Intermediates for Oncology Drug Discovery
The pharmaceutical industry is constantly seeking novel scaffolds that combine multiple bioactive pharmacophores to enhance therapeutic efficacy, particularly in the challenging field of oncology. Patent CN109970753B introduces a groundbreaking methodology for constructing complex xanthone skeleton spliced oxindole or benzofuranone compounds. These molecules represent a sophisticated fusion of two privileged structures: the xanthone core, ubiquitous in natural products like Ergochrome DD, and the spiro-oxindole or spiro-benzofuranone motifs found in drugs such as Satavaptan. This strategic "skeleton splicing" approach creates a new class of poly-functionalized molecules with demonstrated cytotoxicity against human leukemia cells (K562), offering a rich source of chemical entities for high-throughput screening and lead optimization in drug discovery programs.
From a supply chain perspective, the significance of this patent lies not only in the biological potential of the final compounds but also in the robustness of the synthetic route. The ability to access such sterically congested, polycyclic architectures with high stereocontrol is often a bottleneck in medicinal chemistry. By leveraging a specific organocatalytic strategy, this technology bypasses the limitations of traditional transition-metal catalysis, providing a greener, more cost-effective pathway to high-value pharmaceutical intermediates. For R&D teams focused on building diverse libraries for oncology targets, this methodology offers a reliable entry point into chemically space that was previously difficult to explore efficiently.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of spiro-oxindole frameworks fused with other aromatic systems has relied heavily on transition-metal catalysis or harsh acidic/basic conditions. These conventional approaches often suffer from significant drawbacks that hinder their application in large-scale pharmaceutical intermediates manufacturing. Transition metal catalysts, while effective, introduce the risk of heavy metal contamination, necessitating expensive and time-consuming purification steps to meet stringent regulatory limits for residual metals in active pharmaceutical ingredients (APIs). Furthermore, achieving high diastereoselectivity and enantioselectivity simultaneously in the formation of multiple contiguous stereocenters often requires complex ligand design and cryogenic temperatures, drastically increasing operational costs and energy consumption.
Additionally, many traditional methods lack the functional group tolerance required for late-stage diversification. The presence of sensitive moieties often leads to decomposition or side reactions, limiting the scope of substrates that can be utilized. This lack of compatibility restricts the chemical diversity available to medicinal chemists, forcing them to settle for suboptimal analogs or engage in lengthy protection-deprotection sequences. The cumulative effect of these inefficiencies is a prolonged development timeline and inflated cost of goods sold (COGS), which are critical pain points for procurement managers aiming to optimize the budget for early-stage drug discovery projects without compromising on quality or speed.
The Novel Approach
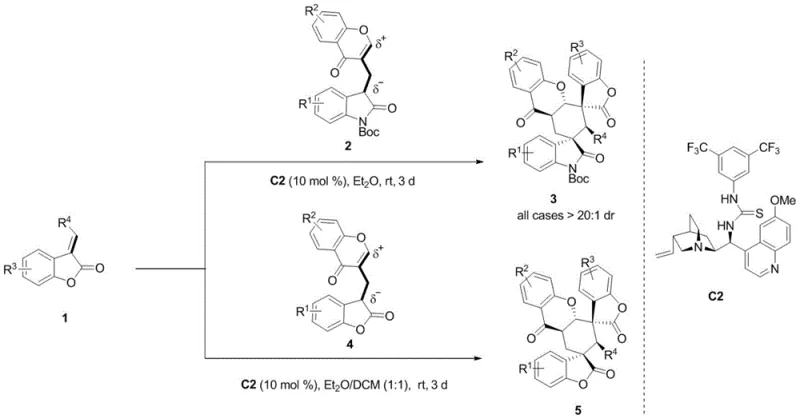
The methodology disclosed in CN109970753B represents a paradigm shift by utilizing a chiral bifunctional organocatalyst to drive a tandem Michael/Michael cycloaddition. This metal-free approach operates under remarkably mild conditions, typically at room temperature in common organic solvents like diethyl ether or dichloromethane. The use of a quinine-derived thiourea catalyst (such as Catalyst C2) enables precise activation of both the nucleophile and the electrophile through hydrogen bonding networks. This dual activation mechanism ensures exceptional stereocontrol, consistently delivering products with enantiomeric excess (ee) values exceeding 99% and diastereomeric ratios (dr) greater than 20:1 across a wide range of substrates.
Beyond the impressive selectivity, this novel route offers substantial advantages in terms of operational simplicity and scalability. The reaction tolerates a broad spectrum of substituents on the aromatic rings, including electron-donating and electron-withdrawing groups like methyl, fluoro, chloro, and trifluoromethyl. This wide substrate scope means that a single optimized protocol can generate a vast library of analogs for structure-activity relationship (SAR) studies without the need for method re-optimization. For supply chain leaders, the elimination of precious metals and the use of stable, commercially available starting materials translate directly into cost reduction in pharmaceutical intermediates manufacturing, ensuring a more resilient and economical supply of these critical building blocks.
Mechanistic Insights into Organocatalytic Michael/Michael Cycloaddition
The success of this transformation hinges on the intricate interplay between the chiral organocatalyst and the reactive substrates. As illustrated in the mechanistic proposal, the thiourea moiety of the catalyst acts as a double hydrogen bond donor, activating the carbonyl group of the chromone synthon. Simultaneously, the basic tertiary amine site of the cinchona alkaloid scaffold deprotonates the acidic methylene group of the oxindole or benzofuranone precursor, generating a nucleophilic enolate. This precise spatial arrangement within the chiral pocket of the catalyst directs the initial Michael addition with high facial selectivity, establishing the first stereocenter with absolute fidelity.
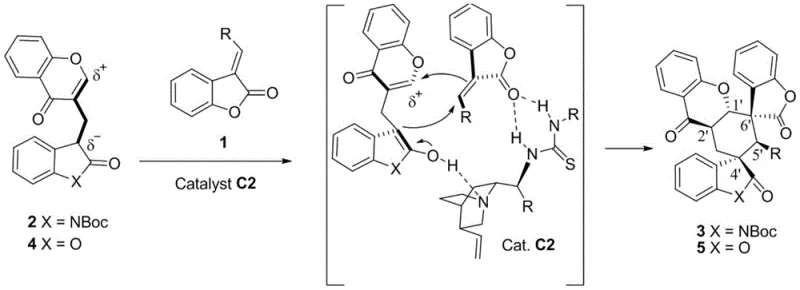
Following the initial carbon-carbon bond formation, the resulting intermediate undergoes a second intramolecular Michael addition to close the spiro-cycle. The rigidity of the catalyst-substrate complex ensures that this cyclization step also proceeds with high stereocontrol, locking in the relative configuration of the newly formed quaternary center. This cascade process effectively constructs a complex hexacyclic framework containing up to four contiguous stereocenters in a single operation. Understanding this mechanism is crucial for R&D directors, as it highlights the robustness of the impurity profile; the high selectivity minimizes the formation of diastereomeric byproducts, thereby simplifying downstream purification and ensuring the delivery of high-purity pharmaceutical intermediates suitable for biological testing.
Furthermore, the compatibility of this mechanism with various solvents and its insensitivity to air and moisture adds another layer of practical value. Unlike sensitive metal complexes that require inert atmosphere gloveboxes, this organocatalytic system can be run in standard reactor setups. This resilience reduces the technical barriers to scale-up, allowing process chemists to transition from milligram-scale discovery to kilogram-scale production with minimal engineering changes. The ability to maintain high yields and selectivity even on larger scales is a testament to the efficiency of the non-covalent interactions driving the reaction, making it an ideal candidate for commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize Xanthone-Spliced Oxindole Compounds Efficiently
The practical execution of this synthesis is designed to be straightforward, aligning with the industry's demand for reproducible and scalable protocols. The general procedure involves dissolving the bifunctional oxindole-chromone synthon and the 3-alkene benzofuranone in a dry organic solvent, followed by the addition of the chiral catalyst. The mixture is then allowed to stir at ambient temperature for a period ranging from 1 to 5 days, depending on the specific electronic nature of the substituents. Reaction progress is monitored via thin-layer chromatography (TLC), and upon completion, the crude product is purified using standard column chromatography techniques to afford the target spiro-compounds as white solids with high optical purity.
- Combine bifunctional oxindole-chromone synthon and 3-alkene benzofuranone in an organic solvent such as diethyl ether.
- Add a chiral bifunctional organocatalyst, specifically a quinine-derived thiourea (e.g., Catalyst C2), at a loading of 10 mol%.
- Stir the reaction mixture at room temperature for approximately 3 days until TLC indicates completion, then purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this organocatalytic technology offers tangible strategic benefits that extend beyond mere chemical elegance. The primary advantage lies in the drastic simplification of the supply chain for raw materials. Since the process relies on organocatalysts derived from abundant natural alkaloids like quinine, rather than scarce and price-volatile precious metals like palladium or rhodium, the risk of supply disruption is significantly mitigated. This stability ensures consistent availability of the catalyst, which is a critical factor in maintaining uninterrupted production schedules for long-term projects.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts removes the need for specialized scavenging resins and extensive metal testing, which are costly and time-consuming steps in API manufacturing. Furthermore, the high stereoselectivity of the reaction minimizes waste generation by avoiding the production of unwanted stereoisomers, thereby improving the overall atom economy. This efficiency translates directly into lower material costs and reduced waste disposal fees, contributing to substantial cost savings over the lifecycle of the drug development program.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions—operating at room temperature and tolerating ambient air—means that the process is less susceptible to variations in utility supplies or equipment failures. This reliability is crucial for meeting tight deadlines in drug discovery. Additionally, the broad substrate scope allows for the rapid synthesis of diverse analogs from a common set of starting materials, reducing lead time for high-purity pharmaceutical intermediates needed for SAR campaigns and enabling faster decision-making in go/no-go milestones.
- Scalability and Environmental Compliance: As regulatory pressure mounts for greener manufacturing processes, this metal-free methodology positions companies favorably regarding environmental compliance. The absence of heavy metals simplifies the environmental impact assessment and reduces the burden of hazardous waste management. Moreover, the simplicity of the work-up procedure facilitates easier scale-up from laboratory to pilot plant, ensuring that the commercial scale-up of complex pharmaceutical intermediates can be achieved without encountering the typical bottlenecks associated with exothermic metal-catalyzed reactions or sensitive reagents.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these xanthone-spliced compounds. These insights are derived directly from the experimental data and background information provided in the patent documentation, aiming to clarify the practical implications for potential partners and licensees.
Q: What is the primary biological activity of these xanthone-oxindole compounds?
A: According to patent CN109970753B, these compounds exhibit significant inhibitory activity against human leukemia cells (K562), indicating strong potential for oncology drug development.
Q: Why is organocatalysis preferred over transition metal catalysis for this synthesis?
A: Organocatalysis eliminates the need for toxic heavy metals, simplifying purification and reducing environmental impact while maintaining excellent stereoselectivity (up to 99% ee).
Q: What are the typical reaction conditions for this transformation?
A: The reaction proceeds under mild conditions, typically at room temperature in solvents like diethyl ether or dichloromethane, requiring about 3 days for completion.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Xanthone-Oxindole Hybrids Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the chemistry described in CN109970753B for advancing oncology research. As a leading CDMO partner, we possess the technical expertise and infrastructure to translate this innovative academic research into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from gram-scale screening to multi-ton manufacturing. We are committed to delivering stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of xanthone-oxindole hybrid meets the highest standards required for preclinical and clinical applications.
We invite you to collaborate with us to leverage this cutting-edge technology for your drug discovery pipeline. Our technical sales team is ready to provide a Customized Cost-Saving Analysis tailored to your specific project needs, demonstrating how our optimized processes can reduce your overall development costs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us help you accelerate the development of next-generation anti-tumor therapeutics.